
[I am an employee of BMS. The views expressed here are my own.]
The blog is long, so I will start with an executive summary. (You can download a pdf copy of the blog here.) Pharmacologic intervention has the opportunity to impact disease progression in the SARS-CoV-2 / COVID-19 crisis. Repurposing of approved therapies is the fastest way to impact patients today, as these medicines have regulatory approval to enable investigator-initiated trials and have a manufacturing process to ensure drug supply. Here, I focus on a specific clinical inflection point in COVID-19 disease progression – hospitalized patients early in their disease course and with signs of a maladaptive immune response, with the intervention intended to prevent disease progression and admission to the ICU. Based on an understanding of disease biology today – which is still quite limited – this clinical inflection point is due to a “maladaptive immune response” seen early in the disease course in patients who later progress to critical illness. Rigorous clinical trials are required to test therapeutic hypotheses related to repurposed therapies, which need to be done in a clinical setting caring for extremely sick patients. Finally, I describe additional research that is required to understand the biology of SARS-CoV-2 / COVID-19, and how such research (e.g.,…
Read full article...

[I am an employee of Bristol-Myers Squibb. The views expressed here are my own.]
One of my predictions for the next decade – the “clear view” decade – is that we will have the ability to click on any gene in the human genome to generate function-phenotype maps. These maps should enable drug discovery by informing on mechanism, magnitude and markers of target perturbation. In particular, I have championed an “allelic series” model, whereby genes with a series of alleles are used to derived genetic dose-response curves (see here, here).
During a recent presentation to my former colleagues at the Division of Genetics at Brigham & Women’s Hospital (BHW, slides here), I discussed important assumptions underlying this model:
- Large-scale sequence data will identify a range of protein-coding variants associated with traits of medical interest that are suitable surrogates for drug discovery (allelic series architecture assumption).
- It will be possible to use high-throughput functional assays to interrogate the impact of trait-associated variants on cell physiology for the majority of genes in the genome (functional readout assumption).
- Large-scale biobanks will emerge to enable testing of these same trait-associated variants for pleiotropic effects across a wide-variety of clinical phenotypes in the real world (PheWAS assumption).
… Read full article...

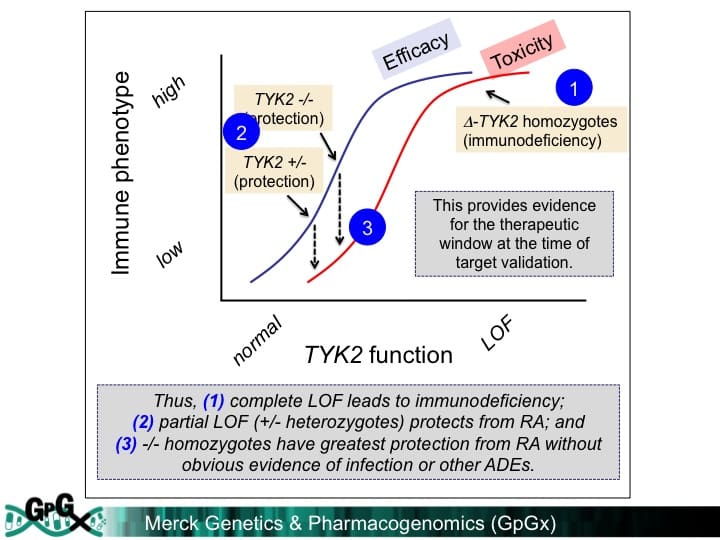
A recent study in the New England Journal of Medicine provides genetic support for a pharmacologically validated target, BAFF, in the treatment of systemic lupus erythematosus. But can human genetics also be used to estimate the target dose and a therapeutic window?
As readers of plengegen.com know, I am constantly on the lookout for published studies that provide insight into the utility of human genetics for drug discovery and development. This past week there was a great post from Francis Collins on the role of the NIH in the discovery (in part via human genetics) and development of tofacitinib (see here), anakinra and potentially novel targets (e.g., STING) for inflammatory diseases (here). Nature Reviews Drug Discovery published a News & Analysis on PCSK9 as a “fertile testing ground for new drug modalities including long-acting RNA interference drugs, vaccines against self-antigens, CRISPR therapeutics and small molecules that control ribosomal activity” (here). New York City released information about a new public health initiative, The NYC Macroscope, which will use electronic health records (EHRs) to track conditions managed by primary care practices that are important to public health..and one day may be linked to genetic data for discovery research (that is me just speculating).…
Read full article...

As readers of my blog know, I am a strong supporter of a disciplined R&D model that focuses on: picking targets based on causal human biology (e.g., genetics); developing molecules that therapeutically recapitulate causal human biology; deploying pharmacodynamic biomarkers that also recapitulate causal human biology; and conducting small clinical proof-of-concept studies to quickly test therapeutic hypotheses (see Figure below). As such, I am constantly on the look-out for literature or news reports to support / refute this model. Each week, I cryptically tweet these reports, and occasionally – like this week – I have the time and energy to write-up the reports in a coherent framework.
Of course, this model is not so easy to follow in the real-world as has been pointed out nicely by Derek Lowe and others (see here). A nice blog this week by Keith Robison (Warp Drive Bio) highlights why drug R&D is so hard.
Here are the studies or news reports from this week that support this model.
(1) Picking targets based on causal human biology: I am a proponent of an “allelic series” model for target identification. Here are a couple of published reports that fit with this model.…
Read full article...

A new sickle cell anemia gene therapy study published in the New England Journal of Medicine (see here, here) gives hope to patients and the concept of rapidly programmable therapeutics based on causal human biology. But how close are we really?
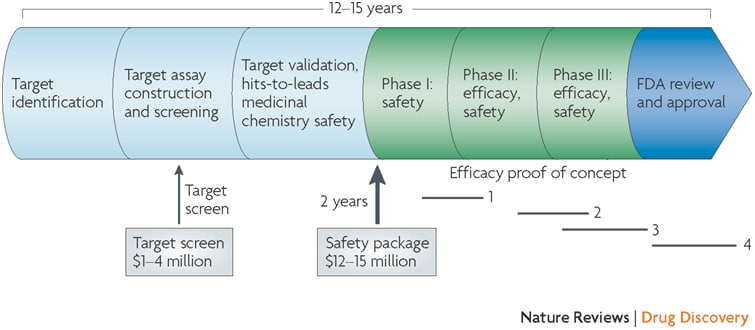
It takes approximately 5-7 years to advance from a therapeutic hypothesis to an early stage clinical trial, and an additional 4-7 years of late stage clinical studies to advance to regulatory approval. This is simply too long, too inefficient and too expensive.
But how can timelines be shortened?
In the current regulatory environment, it is difficult to compress late stage development timelines. This leaves the time between target selection (or “discovery”) and early clinical trials (ideally clinical proof-of-concept, or “PoC”) as an important time to gain efficiencies. Further, discovery to PoC is an important juncture for minimizing failure rates in late development and delivering value to patients in the real world (see here).
Here, I argue that rapidly programmable therapeutics based a molecular understanding of the causal disease process is key to compressing the discovery to PoC timeline.
Imagine a world where the molecular basis of disease is completely understood. For common diseases, germline genetics contributes approximately two-thirds of risk; for rare diseases, germline genetics contributes nearly 100% of risk.…
Read full article...

Inevitably when I post a blog on “human biology” I get a series of comments about the importance of non-human model organisms in drug discovery and development. My position is clear: pick targets based on causal human biology, and then use whatever means necessary to advance a drug discovery program to the clinic.
Very often, non-human model organisms are the “whatever means necessary” to understand mechanism of action. For example, while human genetic studies identified PCSK9 as an important regulator of LDL cholesterol, mouse studies were critical to understand that PCSK9 acts via binding to LDL receptor (LDLR) on the surface of cells (see here). As a consequence, therapeutic antibodies were designed to block circulating PCSK9 from the blood and increase LDLR-mediated removal of circulating LDL (and hopefully to protect from cardiovascular disease).
Moreover, non-human animal models are necessary to understand in vivo pharmacology and safety of therapeutic molecules before advancing into human clinical trials.
Beyond drug discovery, of course, studies from non-human animal models provide fundamental biological insights. Without studies of prokaryotic organisms, for example, we would not have powerful genome-editing tools such as CRISPR-Cas9. Without decades of work on mouse embryonic stem cells, we would not have human induced pluripotent stem cells (iPSCs).…
Read full article...

Today was the second coldest day of my life. When I woke up in Ludlow, Vermont, it was -20 degrees Fahrenheit; with wind chill it was -45° F. As the kids played downstairs, I caught up on my reading comforted by a raging log fire.
The topic de jour: non-genetic examples of causal human biology for drug discovery. Here, the experiment of nature was the formation of autoantibodies against a target and pathway implicated in acquired thrombotic thrombocytopenic purpura (TTP), a life-threatening disorder.
The study that caught my interest, “Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura”, was published last week in the New England Journal of Medicine. I won’t say much about the NEJM article itself, but I will briefly discuss the background leading up to the clinical trial. The key point: autoantibodies against ADAMTS13 pinpointed the target and pathway as causal in the ideal model organism, humans.
The story starts in 1976, when whole blood exchange transfusion resulted in clinical benefit in 8 of 14 patients with TTP. The following year, it was determined that the plasma fraction of the blood was the source of clinical benefit. It took approximately 20 years, however, to identify the deficient plasma factor as ADAMTS13, with deficiency caused by IgG autoantibodies that inhibit the enzyme.…
Read full article...

If you could pick three innovations that would revolutionize drug discovery in the next 10-20 years, what would they be?
I found myself thinking about this question during a recent family vacation to Italy. I was visiting the Galileo Museum, marveling at the state of knowledge during the 1400-1600’s. The debate over planetary orbits seem so obvious now, but the disagreement between church and science led to Galileo’s imprisonment in 1633.
So what is it today that will seem so obvious to our children and grandchildren…and generations beyond? Let me offer a few ideas related to drug discovery, and hope that others will add their own. I am not sure if my ideas are grounded in reality, but that is part of the fun of the game. In addition, “The best way to predict the future is to invent it.”
To start, let me remind readers of this blog that I believe that the three major challenges to efficient drug discovery are picking the right targets, developing the right biomarkers to enable proof-of-concept (POC) studies, and testing therapeutic hypotheses in humans as quickly and safely as possible. Thus, the future needs to address these three challenges.
1.…
Read full article...
The primary purpose of this blog is to recruit clinical scientists into our new Translational Medicine department at Merck (job postings at the end). However, I hope that the content goes beyond a marketing trick and provides substance as to why translational medicine is crucial in drug discovery and development. Moreover, I have embedded recent examples of translational medicine in action, so read on!
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer.]
There is a strong need to recruit clinical scientists into an ecosystem to develop innovative therapies that make a genuine difference in patients. This ecosystem requires those willing to toil away at fundamental biological problems; those committed to converting biological observations into testable therapeutic hypotheses in humans; and those who develop therapies and gain approval from regulatory agencies throughout the world. The first step is largely done in academic settings, and the other two steps largely done in the biopharmaceutical industry…although I am sure there are many who would disagree with this gross generalization!
The term “Translational Medicine” has been broadly used to describe the second step, thereby bridging the Valley of Death between the first and third steps.…
Read full article...
Many of you are probably fully aware of how immuno-oncology is changing cancer treatment. Ken Burns highlighted immunotherapy in his recent PBS series, “Cancer: The Emperor of All Maladies” (video link here). Forbes’ Matthew Herper, BBC and others have written extensively about it, too (here, here). More recently, Genome Magazine had a feature article on the history of immunotherapy (here). As the article states: “The promise of immunotherapy is startling in its simplicity: With a little help from cancer doctors, the patients will cure themselves.”
The key word here is “cure”. Cure!
The purpose of this blog is two-fold: (1) introduce geneticists and genomicists to cancer immunotherapy, if they have not thought about it before, and (2) highlight a recent Science publication by Elaine Mardis, Gerald Linette, and colleagues at WashU (here), with an accompanying News & Views article in Nature (here).
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer.]
Cancer immunotherapy is really cool! As a former practicing rheumatologist at Brigham and Women’s Hospital, I had thought about the role of neoantigens in autoimmunity for many years.…
Read full article...

I admit upfront that this is a self-serving blog, as it promotes a manuscript for which I was directly involved. But I do think it represents a very nice example of the role of human genetics for drug discovery. The concept, which I have discussed before (including my last blog), is that there is a four-step process for progressing from a human genetic discovery to a new target for a drug screen. A slide deck describing these steps and applying them to the findings from the PLoS One manuscript can be found here, which I hope is valuable for those interested in the topic of genetics and drug discovery.
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer. However, the PLoS One study was performed while I was still in academics at BWH/Harvard/Broad.]
Before I provide a summary of the study, I would like to highlight a few recent news stories that highlight that the world thinks this type of information is valuable. First, the state of California is investing US $3-million in a precision medicine project that links genetics and medical records to develop new therapies and diagnostics (here, here).…
Read full article...

Oliver Sacks has terminal cancer. If you have not yet read his heart-warming Op-Ed piece in the New York Times and if you only have five-minutes to spare, then I suggest you read his essay rather than this blog about “experiments of nature” in drug discovery. In his essay, Dr. Sacks concludes with the poignant sentence: “Above all, I have been a sentient being, a thinking animal, on this beautiful planet, and that in itself has been an enormous privilege and adventure.”
Why do I start this week’s blog about articles of the week with this reference? I do so because of two additional – and seemingly unrelated – items from this week: (1) a brief Twitter exchange with David Shaywitz, Derek Lowe, and others that “few people at public biopharmas write interesting stuff, vs consultant-and-PR-driven banality”; and (2) an article in the New York Times Magazine about how Twitter posts can get you into trouble with your employer (see here).
So why do I blog, tweet, etc. given the potential risk? I enjoy the public exchange of ideas because, as Dr. Sacks write, that is the essence of our “sentient being”. I enjoy a network of inter-related ideas for which I can create unique connections.…
Read full article...

Imagine you live in Boston or New York. It is Monday January 26, 2015. You are watching headlines of an impending blizzard, trying to figure out the truth about the weather for the next day. You find that the National Weather Service has a cool online tool – experimental probabilistic snow forecast (see here). As described in Slate magazine (see here), this tool predicted a 67 percent chance of at least 18 inches in New York City.
Unfortunately, most people interpreted this data that there would be 18 inches of snow, not that there could be (with a certain probability) 18 inches of snow.
It was not until Mother Nature did her experiment that we saw the outcome: not much snow in the Big Apple, more than 2 feet of snow in Boston.
The analogy with human genetics is this: it is possible to forecast the functional consequences of deleterious mutations, but it is not until the experimental snow falls – molecular or cellular experiments revealing the functional consequences of mutations – that the functional consequences are actually known. And without knowing the functional consequences of mutations, it is difficult to determine the association of these mutations with human disease.…
Read full article...

Welcome to this second blog post on genetics/genomics for drug discovery! So far, we are 2 for 2. That is, this is the second week in a row where we have reviewed the literature for interesting journal articles and written a blog on why the study is relevant for drug discovery. I say “we”, because this week I asked for input from our Merck Genetic & Pharmacogenomics (GpGx) team. We received a number of interesting submissions from GpGx team members, as summarized at the end of the blog.
This week’s article uses antisense as therapeutic proof-of-concept in humans for a genetic target…again! This story is reminiscent of last week’s post on APOC3 (see here).
Factor XI Antisense Oligonucleotide for Prevention of Venous Thrombosis, New England Journal of Medicine (December 2014).
Summary of the manuscript: While patients with congenital Factor XI deficiency have a reduced risk of venous thromboembolism (VTE), it is unknown whether therapeutic modulation of Factor XI will prevent venous thromboembolism without increasing the risk of bleeding. In this open-label, parallel-group study, 300 patients who were undergoing elective primary unilateral total knee arthroplasty were randomly assigned to receive one of two doses of FXI-ASO (200 mg or 300 mg) or 40 mg of enoxaparin once daily.…
Read full article...

I believe that humans represent the ideal model organism for the development of innovative therapies to improve human health. Experiments of nature (e.g., human genetics) and longitudinal observations in patients with disease can differentiate between cause and consequence, and therefore can overcome fundamental challenges of drug development (e.g., target identification, biomarkers of drug efficacy). Using my Twitter account (@rplenge), this blog (www.plengegen.com/blog), and other forms of social media, I provide compelling examples that illustrate key concepts of “humans as the ideal model organism” (#himo) for drug development.
Why do drugs fail (#whydrugsfail)? This simple question is at the center of problems facing the pharmaceutical industry. In short, drugs fail in early development because of unresolved safety signals or lack of biomarkers for target engagement, and drugs fail in late development because of lack of efficacy or excess toxicity. This leads to a costly system for bringing new drugs to market – not because of the successes, but because >95% of drug programs ultimately fail. Without improvements in rates of success in drug development, the sustainability of the pharmaceutical industry as we know it is in trouble (see here). Not surprisingly, much has been written about this topic, including analyses of development strategies (Forbes blog, Drug Baron), company pipelines (Nature Reviews Drug Discovery manuscript from AstraZeneca) and FDA approvals (here and here).…
Read full article...

At the Spring PGRN meeting last week, there were a number of interesting talks about the need for new databases to foster genetics research. One talk was from Scott Weiss on Gene Insight (see here). I gave a talk about our “RA Responder” Crowdsourcing Challenge (complete slide deck here). Here are a few general thoughts about the databases we need for genomics research.
(1) Silo’s are so last year
Too often, data from one interesting pharmacogenomic study (e.g., GWAS data on treatment response) are completely separate from another dataset that can be used to interpret the data (e.g., RNA-sequencing). Yes, specialized labs that generated the data can integrate the data for their own analysis. And yes, they can release individual datasets into the public for others to stitch together. But is this really what we need? Somehow, we need to make data available in a manner that is fully integrated and interoperable. One simple example of this is GWAS for autoimmune diseases. Since 2006, a large number of genetic data have been published. Still, there is no single place to go see results for all autoimmune diseases, despite the fact that there is tremendous shared overlap among the genetic basis for these diseases.…
Read full article...

I prepared a lecture for immunology graduate students at Harvard Medical School on clinical features of rheumatoid arthritis (RA) for the G1 IMM302qc class.
The slide deck can be found here.
A brief summary:
•Clinical characteristics and pathophysiology
•Differential diagnosis
•Exam and laboratory studies
•Treatment strategy
•Research opportunities
The future research opportunities include using human genetics as an anchor for drug discovery in RA. I briefly go over three strategies:
(1) “look-up” method – simple and suggestive but undisciplined (examples in RA: IL6R/tocilizumab, CTLA4/abatacept)
(2) “Allelic series” method – powerful but likely infrequent (example in other disease: PCSK9)
(3) “pathway” method – powerful and comprehensive but target ID difficult (example in RA: CD40 signaling; Gang Li et al, in press PLoS Genetics)
… Read full article...

This blog post pertains to the Systems Immunology graduate course at Harvard Medical School (Immunology 306qc; see here), which is led by Drs. Christophe Benoist, Nick Haining and Nir Hacohen. My lecture is on the role of human genetics as a tool for understanding the human immune system in health and disease. What follows is an informal description of my lecture. The slide deck for the lecture can be downloaded here. Throughout, I have added key references, with links to the manuscripts and other web-based resources embedded within the blog (and also listed at the end). I highlight five key manuscripts (#1, #2, #3, #4, and #5), which should be reviewed prior to the lecture; the other references, while interesting, are optional.
Overview
It is increasingly clear that humans serve as the best model organism for understanding human health and disease. One reason for this paradigm shift is the lack of fidelity of most animal models to human disease. For systems immunology, the mouse is a powerful model organism to understand fundamental mechanisms of the immune system. However, studies in humans are required to understand how these mechanisms can be translated into new biomarkers and drugs.…
Read full article...

I read with interest a recent publication by Khandpur et al in Science Translational Medicine on NETosis in the pathogenesis of rheumatoid arthritis (download PDF here). It made me think about “cause vs consequence” in scientific discovery. That is, how does one determine whether a biological process observed in patients with active disease is a cause of disease rather than a consequence of disease?
In reading the article, I learned about how neutrophils cause tissue damage and promote autoimmunity through the aberrant formation of neutrophil extracellular traps (NETs). Released via a novel form of cell death called NETosis, NETs consist of a chromatin meshwork decorated with antimicrobial peptides typically present in neutrophil granules. (Read more about NETs on Wikipedia here.)
Mendelian randomization is a method of using measured variation in genes of known function to examine the causal effect of a modifiable exposure on disease in non-experimental studies (read more here). It is a powerful to determine if an observation in patients is causal. For example, if autoantibodies are pathogenic in RA, then DNA variants that influence the formation of autoantibodies should also be associated with risk of RA. This is indeed the case, as exemplified by variants in a gene, PADI4, the codes for an enzyme involved in peptide citrullination (see here). …
Read full article...

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}