
[ I am an employee of Bristol Myers Squibb. The views expressed here are my own, assuming I am real and not a humanoid. ]
In the original Blade Runner (1982), Harrison Ford’s character, Deckard, implements a fictitious Voight-Kampff test to measure bodily functions such as heart rate and pupillary dilation in response to emotionally provocative questions. The purpose: to establish “truth”, i.e., determine whether an individual is a human or a bioengineered humanoid known as a replicant.
While the Voight-Kampff test was used to establish truth for humans vs replicants, the concept of “truth” is central to neural networks used in machine learning and artificial intelligence (AI). And for AI to be effective in drug discovery and development, it is critical to ask a fundamental question: what is “truth” in drug discovery and development?
INTRODUCTION
I recently read the book Genius Makers by Cade Metz and was reminded of the long history of machine learning, neural networks, and artificial intelligence (AI). This is a field more than 60 years in the making, with slow growth for the first 50 years – AI was founded as an academic discipline in 1956 – and exponential growth in the last 10. The original mathematical framework of neural networks was created in the 50’s (perceptron), 60’s and 70’s (backpropagation), but went largely unappreciated outside of academics, as the practical applications were few and far between.…
Read full article...

[I am an employee of Celgene. The views expressed here are my own.]
In the Wizard of Oz, Dorothy clicks her heels and hopes for re-entry from her dream world by repeating, “There’s no place like home…there’s no place like home…” I often feel that many in the genetics community look at their human genetics data with the same youthful optimism as Dorothy – clicking their genetic heels and wishing “my genetic discovery will become a drug…my genetic discovery will become a drug…” But without rigor and discipline, such heel-clicking won’t overcome many of the challenges that face drug hunters along the tortuous journey from a genetic idea to a new medicine.
In this blog, I discuss a recent study on the genetics of multiple sclerosis (MS) published in Science (see here). This is a beautiful study that substantially advances the genetic landscape of patients with a devastating disease. However, the study falls short in terms of the application of human genetics to drug discovery. To chart a course for the future, I introduce the concept of mechanism, magnitude and markers (oh my!), which I refer to as the three M’s. …
Read full article...

[I am an employee of Celgene. All opinions expressed here are my own.]
A meeting was recently convened to discuss a roadmap for understanding the genetics of common diseases (search Twitter for #cdcoxf18). I presented my vision of a genetics dose-response portal (slides here; link to related 2018 ASHG talk here). The organizers (@RachelGLiao, @markmccarthyoxf, @ceclindgren, Rory Collins [Oxford], Judy Cho [New York], @NancyGenetics, @dalygene, @eric_lander) asked participants to share their vision. I thought I would blog about my mine.
You’ll notice my vision is ambitious. Nonetheless, I believe these objectives are feasible to accomplish within a 3-year (Phase 1) and 7-year (Phase 2) time frame. Phase 1 would start immediately and would guide projects for Phase 2. In reality, many aspects of Phase 1 are already underway today (e.g., GWAS catalogue at EBI; Global Alliance for Genomics and Health [GA4GH] data sharing methods). Phase 2 consists of two parts: federation of global biobanks and experimental validation of variants, genes and pathways. Some components of Phase 2 could start today (e.g., exome sequencing in >100,000 cases selected from existing case-control cohorts and biobanks; human knockout project). As with Phase 1, many components of Phase 2 are already underway (federation of existing biobanks [e.g.,…
Read full article...

[I am an employee of Celgene. All views expressed here are my own.]
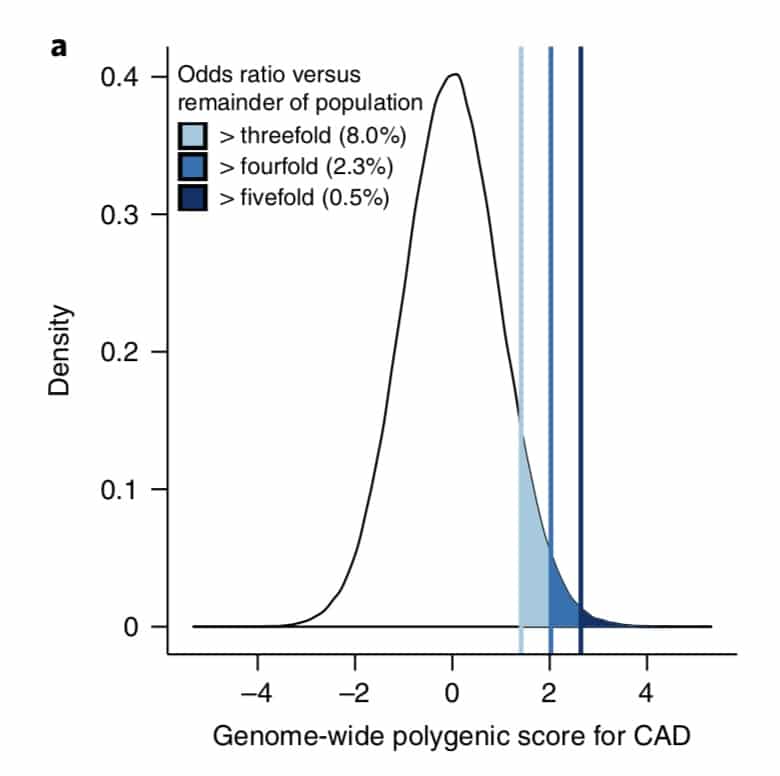
What is the clinical significance of residing within the tail of a distribution for disease risk? A new study published in Nature Genetics uses a composite polygenic score to measure extremes of genetic risk(see original article here). The authors make the bold statement: “it is time to contemplate the inclusion of polygenic risk prediction in clinical care”. In this plengegen.com blog, I briefly review the paper, frame the impact of the study in terms of “long tails”, and propose how genetic tails may be used as part of a healthcare system reimagined.
The premise of the paper is that a genome-wide polygenic score (GPS) – a composite genetic test that includes thousands and sometimes millions of genetic variants – can identify a small number of individuals from the general population that have an elevated risk. The study applies polygenic risk scores to five common diseases but spends most attention to coronary artery disease (CAD). For each disease, the increase in risk is approximately 3- to 5-fold higher among individuals at the extreme of the polygenic tail compared to those in the general population – see Figure 2a (and below) for CAD, where ~8% of the general population is at a 3-fold increase in risk based on a polygenic risk score.…
Read full article...

Over the holidays my family participated in an Escape Room, a live puzzle adventure game. We worked as a team to solve riddles, find clues and, over the course of 60-minutes, complete an old town bank heist. Many of the successful clues came from unexpected places – coordinates on maps, numbers inscribed in hidden places, and physical features of the room itself. Other clues seemed promising, but ultimately led to dead ends. In the end, everything came together and we escaped with only seconds to spare.
And so it goes with the invention of new medicines. The approval of a new medicine is an Escape Room of sorts, but over the course of decades not minutes. And like an Escape Room, clues can come from unexpected places, with some leading to new insights and others leading to dead ends.
I was in an Escape Room state-of-mind as I read a Science Translational Medicine article that developed a system to differentiate blood cells into microglia-like cells to study gene variants implicated in neurodegenerative disorders (here). In this blog, I provide a brief summary of the study, and then describe the potentially interesting phenomenon of genetically driven tissue-specific pathogenicity.…
Read full article...

A new genetics initiative was announced today: the creation of FinnGen (press release here). FinnGen’s goal is to generate sequence and GWAS data on up to 500,000 individuals with linked clinical data and consented for recall. There are many applications for such a resource, including drug discovery and development. In this blog, I want to first describe the application of PheWAS for drug discovery and development, and then introduce FinnGen as a new PheWAS resource (see FinnGen slide deck here).
[Disclaimer: I am an employee of Celgene. The views expressed here are my own.]
PheWAS
PheWAS turns GWAS on its head. While GWAS tests millions of genetic variants for association to a single trait, PheWAS does the opposite: tests hundreds (if not thousands) of traits for association with a single genetic variant. This approach is primarily relevant for those genetic variants with an unambiguous functional consequence – for example, a variant associated with disease risk or a variant that completely abrogates gene function. There are useful online resources (see here), as well as several nice recent reviews by Josh Denny and colleagues, which provide additional background on PheWAS (see here, here).
Work that originated from my academic lab represents the first example of PheWAS for drug discovery – in particular, how to use PheWAS to predict on-target adverse drug events (ADEs) and to select indications for clinical trials (see 2015 PLoS One publication here).…
Read full article...

A new manuscript by Jonathan Pritchard and colleagues published in Cell (see here) has garnered a lot of attention from the genetics community (see here, here, here, here, here). In this blog, I add to the ongoing commentary. I first summarize the main conclusions of the manuscript, and then I discuss the implications for drug discovery and development. For the latter, the three main points are: (1) “core genes” represent good drug targets, especially if they harbor a series of alleles that link function to phenotype; (2) regulatory networks identified by “peripheral genes” point to specific cell types and mechanism that can be used for phenotypic screens; and (3) new approaches are needed to drug cellular networks – what I will refer to as “circuit pharmacology” – as the bulk of drug discovery today is an attempt to reduce complex mechanisms to individual drug targets.
Here is a brief summary of the main conclusions of the manuscript.
- There is a small number of “core genes” that “provide mechanistic insights into disease biology and may suggest druggable targets.” How these core genes are defined, however, remains to be determined. The manuscript suggests a few approaches, including: genes with large effect size variants from GWAS and genes with an allelic series, especially those with lower-frequency variants of larger effects.
… Read full article...

As readers of my blog know, I am a strong supporter of a disciplined R&D model that focuses on: picking targets based on causal human biology (e.g., genetics); developing molecules that therapeutically recapitulate causal human biology; deploying pharmacodynamic biomarkers that also recapitulate causal human biology; and conducting small clinical proof-of-concept studies to quickly test therapeutic hypotheses (see Figure below). As such, I am constantly on the look-out for literature or news reports to support / refute this model. Each week, I cryptically tweet these reports, and occasionally – like this week – I have the time and energy to write-up the reports in a coherent framework.
Of course, this model is not so easy to follow in the real-world as has been pointed out nicely by Derek Lowe and others (see here). A nice blog this week by Keith Robison (Warp Drive Bio) highlights why drug R&D is so hard.
Here are the studies or news reports from this week that support this model.
(1) Picking targets based on causal human biology: I am a proponent of an “allelic series” model for target identification. Here are a couple of published reports that fit with this model.…
Read full article...

Inevitably when I post a blog on “human biology” I get a series of comments about the importance of non-human model organisms in drug discovery and development. My position is clear: pick targets based on causal human biology, and then use whatever means necessary to advance a drug discovery program to the clinic.
Very often, non-human model organisms are the “whatever means necessary” to understand mechanism of action. For example, while human genetic studies identified PCSK9 as an important regulator of LDL cholesterol, mouse studies were critical to understand that PCSK9 acts via binding to LDL receptor (LDLR) on the surface of cells (see here). As a consequence, therapeutic antibodies were designed to block circulating PCSK9 from the blood and increase LDLR-mediated removal of circulating LDL (and hopefully to protect from cardiovascular disease).
Moreover, non-human animal models are necessary to understand in vivo pharmacology and safety of therapeutic molecules before advancing into human clinical trials.
Beyond drug discovery, of course, studies from non-human animal models provide fundamental biological insights. Without studies of prokaryotic organisms, for example, we would not have powerful genome-editing tools such as CRISPR-Cas9. Without decades of work on mouse embryonic stem cells, we would not have human induced pluripotent stem cells (iPSCs).…
Read full article...

I say article of the week, but I have been lazy this summer (or maybe just consumed by other things). My last “article of the week” was in May and my last Plengegen blog post was over a month ago!
By now everyone knows the PCSK9 story. Human genetics identified the target; functional work in mouse and human cells led to a mechanistic understanding of PCSK9’s role in LDL receptor recycling; therapeutic modulation was shown to lower LDL cholesterol in clinical trials; and the FDA approved drugs based on LDL lowering, with outcome trials underway to demonstrate (presumably) cardiovascular benefit. What the story highlights is that a mechanistic understanding of causal pathways in human disease is key to the success of translating targets into therapies. Further, the PCSK9 story underscores the importance of a simple biomarker (LDL cholesterol) to measure a complex causal pathway in a clinical trial.
A recent study in the New England Journal of Medicine (NEJM) provides insight into a putative causal pathway in obesity, and thus a potentially a new mechanism for therapeutic modulation. The accompanying Editorial also provides a nice perspective.
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer.…
Read full article...

I admit upfront that this is a self-serving blog, as it promotes a manuscript for which I was directly involved. But I do think it represents a very nice example of the role of human genetics for drug discovery. The concept, which I have discussed before (including my last blog), is that there is a four-step process for progressing from a human genetic discovery to a new target for a drug screen. A slide deck describing these steps and applying them to the findings from the PLoS One manuscript can be found here, which I hope is valuable for those interested in the topic of genetics and drug discovery.
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer. However, the PLoS One study was performed while I was still in academics at BWH/Harvard/Broad.]
Before I provide a summary of the study, I would like to highlight a few recent news stories that highlight that the world thinks this type of information is valuable. First, the state of California is investing US $3-million in a precision medicine project that links genetics and medical records to develop new therapies and diagnostics (here, here).…
Read full article...

There was an eruption in Iceland last week. No, this was not another volcanic eruption. Rather, there was a seismic release of human genetic data that provides a glimpse into the future of drug discovery. The studies were published in Nature Genetics (the issue’s Table of Contents can be found here), with insightful commentary from Carl Zimmer / New York Times (here), Matthew Herper / Forbes (here), and others (here, here).
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer.]
As I have commented before, human genetics represent a very powerful approach to identify new drug targets (see here, here). I have articulated a 4-step process (see slide #5 from this deck): (1) select a phenotype that is relevant for drug discovery; (2) identify a series of genetic variants (or “alleles”) that is associated with the phenotype; (3) assess the biological function of phenotype-associated alleles; and (4) determine if those same alleles are associated with other phenotypes that may be considered adverse drug events.
There is an important assumption about this model: genes with an “allelic series” will be identified from large-scale genetic studies, and these phenotype-associated alleles will serve as an estimate of function-phenotype dose-response curves.…
Read full article...
This week’s theme is genes to function for drug screens…with a macabre theme of zombies! As more genes are discovered through GWAS and large-scale sequencing in humans, there is a pressing need to understand function. There are at least two steps: (1) fine-mapping the most likely causal genes and causal variants; and (2) functional interrogation of causal genes and causal variants to move towards a better understanding of causal human biology for drug screens (“from genes to screens”).
Genome-editing represents one very powerful tool, and the latest article from the laboratory of Feng Zhang at the Broad Institute takes genome-editing to a new level (see Genetic Engineering & Biotechnology News commentary here). They engineer the dead!
Genome-scale gene activation by an engineered CRISPR-Cas9 complex, Nature (December 2014).
Since its introduction in late 2012, the CRISPR-Cas9 gene-editing technology has revolutionized the ways scientists can apply to interrogate gene functions. Using a catalytically inactive Cas9 protein (dead Cas9, dCas9) tethered to an engineered single-guide RNA (sgRNA) molecule, the authors demonstrated the ability to conduct robust gain-of-function genetic screens through programmable, targeted gene activation.
Earlier this year, the laboratories of Stanley Qi, Jonathan Weissman and others \ reported the use of dCas9 conjugated with a transcriptional activator for gene activation (see Cell paper here).…
Read full article...

I have come across three reports in the last few days that help me think about the question: How many genomes is enough? My conclusion – we need a lot! Here are some thoughts and objective data that support this conclusion.
(1) Clinical sequencing for rare disease – JAMA reported compelling evidence that exome sequencing identified a molecular diagnosis for patients (Editorial here). One study investigated 2000 consecutive patients who had exome sequencing at one academic medical center over 2 years (here). Another study investigated 814 consecutive pediatric patients over 2.5 years (here). Both groups report that ~25% of patients were “solved” by exome sequencing. All patients had a rare clinical presentation that strongly suggested a genetic etiology.
(2) Inactivating NPC1L1 mutations protect from coronary heart diease – NEJM reported an exome sequencing study in ~22,000 case-control samples to search for coronary heart disease (CHD) genes, with follow-up of a specific inactivating mutation (p.Arg406X in the gene NPC1L1) in ~91,000 case-control samples (here). The data suggest that naturally occurring mutations that disrupt NPC1L1 function are associated with reduced LDL cholesterol levels and reduced risk of CHD. The statistics were not overwhelming despite the large sample size (P=0.008, OR=0.47). …
Read full article...

I prepared a lecture for immunology graduate students at Harvard Medical School on clinical features of rheumatoid arthritis (RA) for the G1 IMM302qc class.
The slide deck can be found here.
A brief summary:
•Clinical characteristics and pathophysiology
•Differential diagnosis
•Exam and laboratory studies
•Treatment strategy
•Research opportunities
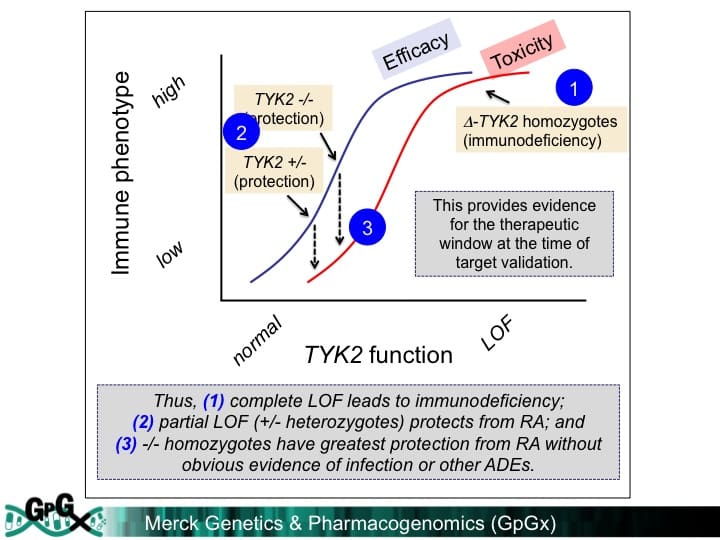
The future research opportunities include using human genetics as an anchor for drug discovery in RA. I briefly go over three strategies:
(1) “look-up” method – simple and suggestive but undisciplined (examples in RA: IL6R/tocilizumab, CTLA4/abatacept)
(2) “Allelic series” method – powerful but likely infrequent (example in other disease: PCSK9)
(3) “pathway” method – powerful and comprehensive but target ID difficult (example in RA: CD40 signaling; Gang Li et al, in press PLoS Genetics)
… Read full article...

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}