
As readers of my blog know, I am a strong supporter of a disciplined R&D model that focuses on: picking targets based on causal human biology (e.g., genetics); developing molecules that therapeutically recapitulate causal human biology; deploying pharmacodynamic biomarkers that also recapitulate causal human biology; and conducting small clinical proof-of-concept studies to quickly test therapeutic hypotheses (see Figure below). As such, I am constantly on the look-out for literature or news reports to support / refute this model. Each week, I cryptically tweet these reports, and occasionally – like this week – I have the time and energy to write-up the reports in a coherent framework.
Of course, this model is not so easy to follow in the real-world as has been pointed out nicely by Derek Lowe and others (see here). A nice blog this week by Keith Robison (Warp Drive Bio) highlights why drug R&D is so hard.
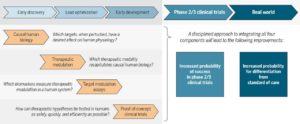
Here are the studies or news reports from this week that support this model.
(1) Picking targets based on causal human biology: I am a proponent of an “allelic series” model for target identification. Here are a couple of published reports that fit with this model.
• AKT2 and type 2 diabetes. Published in the journal Diabetes (here), Cecilia Lindgren, Anna Gloyn, Mark McCarthy, and colleagues identify a novel association between an AKT2 loss-of-function (LoF) coding variant (p.Pro50Thr) and increased fasting plasma insulin levels, decreased insulin sensitivity, and increased risk of type 2 diabetes (odds ratio = 1.05). The same AKT2 gene harbors rare, fully penetrant mutations that cause monogenic glycemic disorders. Based on these genetic data, a therapeutic hypothesis is: agonize AKT2 or the pathway to stimulate insulin release and improve glycemic control in diabetes. Is this possible? I am not sure, but certainly human genetics would support a good look into the mechanism.
• ANGPTL3 and coronary artery disease. Published in the Journal of the American College of Cardiology (here, Editorial here), Sekar Kathiresan and colleagues provide three lines of evidence that inhibiting ANGPTL3 will protect against risk of coronary artery disease. First, 3 compound heterozygous members of a family segregating ANGPTL3 LoF mutations had no evidence of coronary atherosclerotic plaque based on computed tomography angiography. Second, heterozygous carriers of ANGPTL3 LoF mutations demonstrated a reduction in circulating triglycerides, LDL cholesterol and risk of coronary artery disease. Third, individuals with lower levels of circulating ANGPTL3 protein had reduced odds of suffering a myocardial infarction.
(2) Developing molecules that therapeutically recapitulate causal human biology: Increasingly, I am convinced this is the major bottleneck to an efficient R&D pipeline. As more data emerge from humans to pick causal targets and pathways (as above), the ability to manipulate those targets with a therapeutic molecule will become rate-limiting. This perspective is the reason for my increased interest in “programmable therapeutics” and related topics such as “drugging the undruggable”.
- • Protein-degradation technology. Craig Crews published a review in Science describing an approach which he calls “event-base pharmacology” (here, see figure below). I am intrigued by proteolysis-targeting chimera (PROTAC) technology, as it has the potential (underscore potential!) to become rapidly recursive (i.e., programmable). Take a look at the review and let your imagination drift towards a world where (a) every intracellular target is tagged by a small molecule, (b) each of the >700 E3 ligases is tagged by a small molecule ligand, and (c) pairs of each can be linked together and introduced quickly into humans for proof-of-concept studies. Hmmmm….
- • Warp Drive Bio partnership with GSK. The Warp Drive Bio website proudly declares that their mission is to exploit “Molecules and Mechanisms of Nature” to create transformative medicines. As described in a nifty corporate slide deck, Warp Drive wants to extend the therapeutic mechanism of rapamycin to other targets. While still early, such bold efforts to drug the undruggable are desperately needed. This week, Warp Drive is one step closer to their goal, with a partnership with GSK.
- • Target deconvolution for PCSK9 phenotypic screen. As my grandmother used to tell me, “If you can’t drug it, turn it into a phenotypic screen.” Published in PLoS Biology (here), scientists from Pfizer describe how they deconvoluted a previously published phenotypic screen. Unexpectedly, the target of the PCSK9-lowering small molecule appears to be a PCSK9-specific ribosome elongation complex, although additional studies are required to sort through the details. (In case you are still reading: my grandmother never actually said that.)
- • Fragment-based phenotypic screens. As phenotypic screens increase in popularity, new approaches are needed to chemically probe relevant assays. A News & Analysis in Nature Reviews Drug Discovery (here) provides a perspective on a Cell paper from January (here) and a new start-up company, Vividion Therapeutics (here). Cool stuff.
- • Ionis subsidiary Akcea plans for IPO. I put this here, as the foundation of Ionis is anti-sense oligonucleotide (ASO) technology. Most of their targets are rooted in human genetics, including two highlighted in John Carroll’s Endpoints story [e.g., APOC3, Lp(a)]. Nucleic acid-based therapeutics continue their resurgence, although still have many challenges to overcome. As below, ASO’s are being developed for a genetic target in ALS.

(3) Deploying pharmacodynamic biomarkers that also recapitulate causal human biology: The ideal pharmacodynamic (PD) biomarker should be linked to a target via the same causal mechanism that implicates the target itself in human disease. Unfortunately, many biomarkers used in clinical trials represent downstream measurements that may or may not be directly correlated with target perturbation. This week, there was an impressive study linking a specific mutation with a circulating protein in a rare disease, amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease).
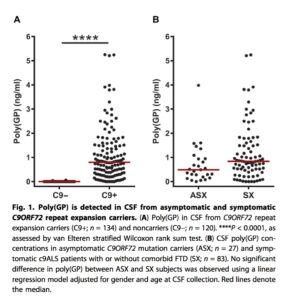
• PD biomarker in ALS. A study in Science Translational Medicine describes how poly(GP) proteins, translated from the mysterious C9ORF72 gene, are stably detected in the cerebral spinal fluid (CSF) in mutation carriers (both asymptomatic and patients with ALS; see figure below). They go on to show that an ASO directed against the RNA product of the C9ORF72 gene reduced poly(GP) protein levels in two in vitro human cell systems (lymphoblastoid and iPSC-derived neurons) and an in vivo mouse model of ALS. If a drug is developed based on this genetic target and PD biomarkers such as poly(GP) protein, it will represent an example of what I predict will be the future of “precision medicine” (see previous blog here).
One note of caution related to the C9ORF72 mutation: it is not clear if the underlying molecular defect is gain- or loss-of-function. The mutation itself is a hexanucleotide repeat expansion, where normal individuals have <30 repeats and patients with ALS or a related condition, frontotemporal dementia (FTD), have hundreds to thousands of repeats. There are two major theories about the way that the C9ORF72 mutation causes disease: (1) gain-of-function: accumulation of intracellular RNA becomes toxic; or (2) loss-of-function: haploinsufficiency of C9ORF72 protein somehow leads to disease. The poly(GP) protein biomarker would only be effective if the molecular mechanism is gain-of-function.
(4) Conducting small clinical proof-of-concept studies to quickly test therapeutic hypotheses: I am using a bit of poetic license for this category. Strictly speaking, this category should be reserved for small, early development clinical trials that demonstrate, for the first time, that a molecule is effective in humans. And by effective, I don’t just mean safe and tolerable or that a downstream and potentially irrelevant biomarker changes levels. By effective, I mean that either it changes a proximal / causal biomarker [as in poly(GP) protein levels in ALS – see above] or it makes patients clinically better. Here, however, I am highlighting medicines that were reported to demonstrate clinical efficacy in Phase III (tezacaftor/ivacaftor) were approved by the FDA (ocrelizumab, dupilumab).
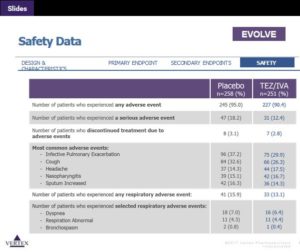
- • Combination therapy in cystic fibrosis. This was big news, with several well-written summaries by real journalists: Matthew Herper (here), John Carroll (here), and Adam Feuerstein (here). Clearly the target, the CFTR protein, is a validated human genetic target. What is most relevant here is that the combination drug is safe (see Table below), setting the stage for future triple combinations for improved efficacy. In terms of small, clinical PoC pipeline, the cystic fibrosis field now has a clear path: change in FEV1 at 2 weeks is a strong predictor of clinical utility (see Figure 1 in Ramsey et al NEJM 2011).
- • Anti-IL4Ra in atopic dermatitis. This week, the FDA approved Dupixent (dupilumab), an injectable biologic for adults with moderate-to-severe eczema (atopic dermatitis). The drug is an antibody that binds to interleukin-4 receptor alpha subunit (IL-4Ra), which in turn modulates signaling of both the IL4 and IL13 pathways. Human genetic studies have identified the IL13 axis as important in atopic dermatitis (see 2015 Nature Genetics study here). Thus, while human genetics did not drive the discovery and early development of dupilumab, this is a nice retrospective example of the value of human genetics for drug discovery and development. As with CF, there is also a clear path to clinical PoC in small, early development trials: change in eczema severity or pruritus at 4 weeks predicts clinical utility (see Figure 1 in Beck et al NEJM 2014).
- • Anti-CD20 therapy in multiple sclerosis (MS). According to a wonderful STAT article by Ron Winslow, the FDA approval of Ocrevus (ocrelizumab) represents a 40-year journey. The drug is a modern-day version of rituximab: a humanized anti-CD20 monoclonal antibody that targets CD20 marker on B lymphocytes. It is now approved for both Relapse-Remitting Multiple Sclerosis (RRMS) and Primary-Progressive Multiple Sclerosis (PPMS). It is the first and only FDA-approved treatment for PPMS. Human genetics implicates B and T lymphocytes in the pathogenesis of MS, although the signal is stronger for T-helper cells than other cell types (see Nature 2011). CD20 itself has not emerged as a gene directly implicated in MS pathogenesis via genetics. Other experimental observations in humans (e.g., B cell receptor diversity in the peripheral compartments and the CSF of patients with multiple sclerosis), however, provide a concrete link between B cells and MS (see Dendrou et al Nature Reviews Immunology 2015).
That’s all, folks. If you have examples that support or refute a disciplined model of R&D as described here, please send them my way.