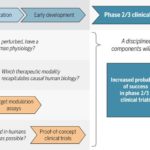
As readers of my blog know, I am a strong supporter of a disciplined R&D model that focuses on: picking targets based on causal human biology (e.g., genetics); developing molecules that therapeutically recapitulate causal human biology; deploying pharmacodynamic biomarkers that also recapitulate causal human biology; and conducting small clinical proof-of-concept studies to quickly test therapeutic hypotheses (see Figure below).As such, I am constantly on the look-out for literature or news reports to support / refute this model.Each week, I cryptically tweet these reports, and occasionally – like this week – I have the time and energy to write-up the reports in a coherent framework.
Of course, this model is not so easy to follow in the real-world as has been pointed out nicely by Derek Lowe and others (see here).A nice blog this week by Keith Robison (Warp Drive Bio) highlights why drug R&D is so hard.
Here are the studies or news reports from this week that support this model.
(1) Picking targets based on causal human biology: I am a proponent of an “allelic series” model for target identification. Here are a couple of published reports that fit with this model.…
Like many, I waited with bated breath for results of the anti-PCSK9 (evolocumab) FOURIER cardiovascular outcome study last week. There have been many interesting commentaries written on the findings.A few of my favorites are listed here (Matthew Herper), here (David Grainger), here (Derek Lowe), and here (Larry Husten), amongst others, with summaries provided at the end of this blog.Most of these articles focused on clinical risk reduction vs. what was predicted for cardiovascular outcome, as well as whether payers will cover the cost of the drugs.These are incredibly important topics, and I won’t comment on them further here, other than to say that the debate is now about who should get the drug and how much it should cost.
In this blog, I want to emphasize key points that pertain to human genetics and drug discovery.And make no mistake: the anti-PCSK9 story and FOURIER clinical trial outcome is a triumph for genetics and drug discovery. This message seems to be getting muddled, however, given the current cost of evolocumab and the observation that cardiovascular risk reduction was less than expected, based on predictions from a 2005 study published by Cholesterol Treatment Trialists (CTT) (see Lancet study here).…
Inevitably when I post a blog on “human biology” I get a series of comments about the importance of non-human model organisms in drug discovery and development. My position is clear: pick targets based on causal human biology, and then use whatever means necessary to advance a drug discovery program to the clinic.
Very often, non-human model organisms are the “whatever means necessary” to understand mechanism of action. For example, while human genetic studies identified PCSK9 as an important regulator of LDL cholesterol, mouse studies were critical to understand that PCSK9 acts via binding to LDL receptor (LDLR) on the surface of cells (see here). As a consequence, therapeutic antibodies were designed to block circulating PCSK9 from the blood and increase LDLR-mediated removal of circulating LDL (and hopefully to protect from cardiovascular disease).
Moreover, non-human animal models are necessary to understand in vivo pharmacology and safety of therapeutic molecules before advancing into human clinical trials.
Beyond drug discovery, of course, studies from non-human animal models provide fundamental biological insights. Without studies of prokaryotic organisms, for example, we would not have powerful genome-editing tools such as CRISPR-Cas9. Without decades of work on mouse embryonic stem cells, we would not have human induced pluripotent stem cells (iPSCs).…
At the Harvard-Partners Personalized Medicine Conference last week I participated in a panel discussion on complex traits. When asked about where personalized medicine for complex traits will be in the future, I answered that I envision two major categories for personalized therapies.
(1)Development of drugs based on genetic targets will lead to personalized medicine; and
(2)Large effect size variants will be detected in clinical trials or in post-approval studies and will lead to personalized medicine.
This answer, I said, was based in part on current categories of FDA pharmacogenetic labels and in part on how I see new drug discovery occurring in the future. But did the current FDA labels really support this view?
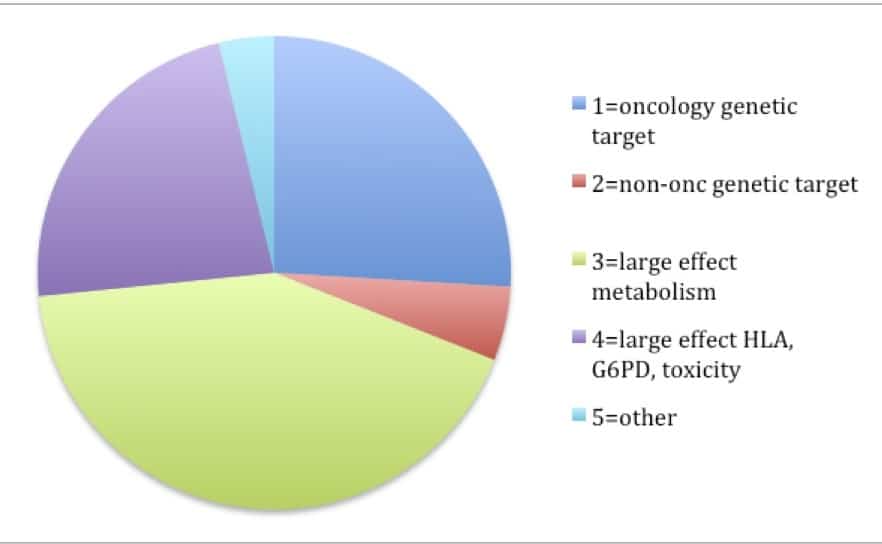
The answer is “yes”. In reviewing the 158 FDA labels (Excel spreadsheet here), my crude analysis found that 31% of labels fall into the “genetic target” category (most from oncology – 26% of total) and 65% fall into the “large effect” category (most from drug metabolism [42% of total], HLA or G6PD [15% of total]).
A subtle but important point is that I predict that category #2 (PGx markers for non-oncology “genetic targets”) will grow in the future. In other words, development of non-oncology drugs will riff-off the success of drugs developed based on somatic cell genetics in oncology. …
So, you have a target and want to start a drug discovery program, do ya? How would you do it?
When I was at Brigham and Women’s Hospital, Harvard Medical School and the Broad Institute, I presented an idea from an early GWAS of rheumatoid arthritis (RA, see here) to Ed Scolnick (former president of Merck Research Labs, now founding director of the Stanely Center at the Broad Institute, see here). In this study, we found evidence that a non-coding variant at the CD40 gene locus increased risk of RA. The first questions he asked: How does the genetic mutation alter CD40 function? Is it gain-of-function or loss-of-function? What assay would you use for a high-throughput small molecule screen to recapitulate the genetic finding?
I was caught off-guard. Sadly, I had never really thought about all of the details. At the time, I knew enough as a clinician, biologist and a geneticist to appreciate that CD40 was an attractive drug target for RA. However, I was quite naïve to the steps required to take a target into a drug screen. That simple conversation led to several years worth of work, which ultimately led to a proof-of-concept phenotypic screen published in PLoS Genetics five years later (see here).…
Question: What can we learn from Sputnik (see here), DARPA (see here) and disruptive innovation (see here) to invent new drugs?
Answer: The best way to prevent surprise is to create it. And if you don’t create the surprise, someone else will. (This is a cryptic answer, I know, but I hope the answer will become clearer by the end of the blog.)
My previous blogs highlighted (1) the pressing need to match an innovative R&D culture with an innovative R&D strategy rooted in basic science (see here), and (2) the importance of phenotype in target ID and validation (TIDVAL) efforts anchored in human genetics (see here). Now, I want to flesh out more of the scientific strategy around human genetics – with a focus on single genes and single drug targets.
To start, I want to frame the problem using an unexpected source of innovation: the US government.
There is an interesting article in Harvard Business Review on DARPA and “Pasteur’s Quadrant” – use-inspired, basic-science research (see here and here). This theme is critically important for drug discovery, as the biopharma industry has a profound responsibility to identify new targets with increased probability-of-success and unambiguous promotable advantage (see here). …
As I sought advice from colleagues about my career, I was frequently asked if I would prefer to work in academics or industry (emphasis on the word “or”). The standard discussion went something like this:
ACADEMICS – you are your own boss and you are free to chose your own scientific direction; funding is tight, but good science still gets funded by the NIH, foundations and other organizations (including industry); the team unit centers around individuals (graduate students, post-docs, etc), which favors innovative science but sometimes makes large, multi-disciplinary projects challenging; there is long-term stability, including control over where you want to work and live, assuming funding is procured and good ideas continue; your base salary will be less than in industry, but you still make a good living and there are opportunities to consult – and maybe even start your own company – to supplement income. Bottom line: if you want to do innovative science under your own control, work in academics – as that is where most fundamental discoveries are made.
INDUSTRY – there are more resources, but those resources are not necessarily under your control (depending upon your seniority); the company may change direction quickly, which changes what you are able to work on; while drug development takes 10-plus years, many goals are short-term (several years), which limits long-term investment in projects that are risky and require years to develop; the team unit centers around projects (e.g.,…
Bill James developed the “Keltner list” to serve as a series of gut-check questions to test a baseball player’s suitability for the Hall of Fame (see here). The list comprises 15 questions designed to aid in the thought process, where each question is designed to be relatively easy to answer. As a subjective method, the Keltner list is not designed to yield an undeniable answer about a player’s worthiness. Says James: “You can’t total up the score and say that everybody who is at eight or above should be in, or anything like that.”
The Keltner list concept has been adapted to address to serve as a common sense assessment of non-baseball events, including political scandals (see here) and rock bands like Devo (see here).
Here, I try out this concept for genetics and drug discovery. That is, I ask a series of question designed to answer the question: “Would a drug against the product of this gene be a useful drug?” I use PCSK9 as one of the best examples (see brief PCSK9 slide deck here). I also used in on our recent study of CD40 in rheumatoid arthritis, published in PLoS Genetics (see here).…






{kind=link}
{kind=link}
{kind=link}
{kind=link}