

[I am an employee of Celgene. The views expressed here are my own.]
In the Wizard of Oz, Dorothy clicks her heels and hopes for re-entry from her dream world by repeating, “There’s no place like home…there’s no place like home…” I often feel that many in the genetics community look at their human genetics data with the same youthful optimism as Dorothy – clicking their genetic heels and wishing “my genetic discovery will become a drug…my genetic discovery will become a drug…” But without rigor and discipline, such heel-clicking won’t overcome many of the challenges that face drug hunters along the tortuous journey from a genetic idea to a new medicine.
In this blog, I discuss a recent study on the genetics of multiple sclerosis (MS) published in Science (see here). This is a beautiful study that substantially advances the genetic landscape of patients with a devastating disease. However, the study falls short in terms of the application of human genetics to drug discovery. To chart a course for the future, I introduce the concept of mechanism, magnitude and markers (oh my!), which I refer to as the three M’s. The 3M’s concept highlights the challenges and potential solutions of progressing a genetics idea to a therapeutic hypothesis and eventually (click heels now) a new medicine. I discuss the application of this 3M’s framework as it pertains to an allelic series model of drug discovery.
Summary of MS genetics study
The study, which was published in Science (link here), includes genotype data from 47,429 MS cases and 68,374 control subjects – by far the largest genetic study in MS to date. The study identifies 233 statistically independent, genome-wide significant associations with MS susceptibility: 32 within the MHC, 1 on the X chromosome, and 200 on autosomes but outside of the MHC. Further, they identify 416 variants that had evidence of statistical replication but did not reach the level of genome-wide statistical significance. By integrating gene expression and epigenetic data, these findings implicate subsets of immune cells (B cells, T cells, NK cells, microglia, myeloid cells) – but not astrocytes or neurons – as contributing to the earliest events that trigger MS. They demonstrate that approximately half of the MS-associated variants are cis-eQTLs in either cortical neurons or subsets of immune cells (CD4+ T cells, monocytes, PBMCs).
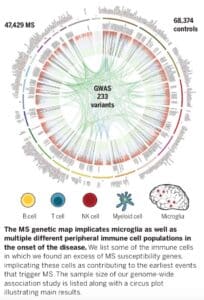
The study highlights a number of challenges faced by drug hunters who want to generate new therapeutic hypotheses from large-scale human genetic studies. The first is the “genetic architecture” challenge: MS is highly polygenic with small effect sizes for nearly all of the implicated regions across the genome. Here are some numbers that provide context:
• 26,395 SNPs reached genome-wide significance (P< 5×10-8) and another 576,204 SNPs had at least nominal evidence of association (5×10-8 > P < 0.05).
• From this long list of variants, genomic partitioning identified 1,961 non-MHC autosomal regions that included 4,842 presumably statistically independent SNPs.
• 9% of regions (31 out of 156) harbored more than one statistically independent GW effect.
• The odds ratios (ORs) of these genome-wide effects ranged from 1.06 to 2.06
A model that includes the extended MHC region and genome-wide significant variants outside of the MHC can explain ~39% of the genetic predisposition to MS, which can be expanded to ~48% if suggestive effect variants are included in the model.
The second challenge is the “functional follow-up” challenge: for each associated variant, there is linkage disequilibrium among the index variant and other variants in the region, with most of these variants (and therefore the likely causal variant) landing outside of protein-coding sequences. As a consequence, it is very difficult to pinpoint which of the associated variants are likely driving the disease association.
To address the functional follow-up challenge, the IMSGC study provides state-of-the-art analyses to extract functional information, including:
-
- • Enrichment of gene expression and epigenetic features with the 200 non-MHC autosomal variants to prioritize potential pathogenic cell types and tissues.
• Enrichment was observed for immune cells that have long been studied in MS, e.g. T cells, as well as in other immune cells such as B cells, innate immune cells (natural killer cells, dendritic cells), and resident immune cells in the CNS (e.g., microglia).
• Importantly, enrichment for MS genes was not observed for astrocytes or neurons.
-
- • Co-localization of variants that control expression of nearby genes (i.e., expression quantitative trait loci, or eQTLs) with variants that influence risk of MS.
• Multiple cell types were interrogated: naive CD4+ T cells and monocytes from 211 healthy subjects; peripheral blood mononuclear cells (PBMCs) from 225 remitting relapsing MS subjects; and dorsolateral prefrontal cortex from two longitudinal cohort studies of aging (n=455 cognitively non-impaired individuals).
• Over the CNS and the three immune sets of data, 104 genome-wide significant MS risk variants co-localized with cis-eQTLs for 203 unique genes, with several appearing to be specific for one of the cell/tissue types.
-
- • Pathway-based analysis based on prioritized genes from the 200 non-MHC autosomal variants.
• Using a genomic-features approach genomic-features approach to nominate most likely causal genes in the region, they prioritized 551 candidate MS genes to test for statistical enrichment of known pathways.
• This approach suggests biological process associated with MS risk, including processes of development, maturation, and terminal differentiation of T cells, B cells, dendritic cells and natural killer cells.
• A protein-protein interaction analysis using GeNets demonstrated that 551 prioritized genes (n=190; 34.5%) were connected and organized into 13 communities, i.e. sub-networks with higher connectivity (p-value: < 0.002).
The three M’s of target perturbation
While the IMSGC publication is a beautiful study that represents the bleeding-edge of genome science, it falls short of what is required to nominate novel drug targets. This criticism is not unique to the IMSGC study – indeed, it is true for nearly every large-scale GWAS published to date. To address this shortcoming, I introduce a concept, the 3Ms of target perturbation: mechanism, magnitude, and markers. Any genetic study should strive to address these 3M’s when nominating a new target based on human genetics.
Mechanism refers to the molecular mechanism of the trait-associated variant (e.g., protein-truncating variant that abolishes protein function, non-coding variant that changes gene expression) and therefore the mechanism by which to perturb therapeutically a target to achieve clinical benefit.
Magnitude refers to the amount by which a trait-associated variant alters the biology of the target (e.g., 50% increase in gene expression, complete null knockout) and therefore the magnitude by which to perturb therapeutically a target to achieve clinical benefit.
Markers refers to translational biomarkers that can be linked to molecular mechanism and measured in a clinical trial to quantitatively assess the magnitude of therapeutic perturbation in small, proof-of-biology clinical trial. In other words, functional studies of trait-associated variants should have the explicit goal of delivering data that informs on the 3 M’s (mechanism, magnitude, and markers).
[Note that there is a 4th M – modality – that is also important. However, I embedded this M within “mechanism”, as the therapeutic modality must be matched to the molecular mechanism of the trait-associated genetic variants. That, and a fourth M would interfere with my Wizard of Oz analogy!]
There are recent examples of approved therapies that reinforce the importance of this 3M framework for genetic targets. Nusinersen (Spinraza) was approved for spinal muscular atrophy; the mechanism of target modulation is through an alteration of gene splicing. Tezacaftor/ivacaftor (Symdeko) was approved for combination therapy in cystic fibrosis; the magnitude of target modulation is ~25% restoration of CFTR levels / function. Evolocumab (Repatha) and alirocumab (Praluent) were approved for hypercholesterolemia; the marker for proof-of-biology is LDL cholesterol.
A path forward for MS and other common diseases
What are the options to address the 3 M’s of target perturbation and therefore nominate new drug targets for MS? Here, I lay out one possible approach, which is part of a broader discussion within the genetics community (see here for International Common Disease Alliance [ICDA], which launched this week; link to white paper here). The approach described here builds on the “allelic series” framework that I have described previously (here, here, here). The current IMSGC Science study represents progress towards the first four steps, as is true for many high-quality genetic studies. As I describe, however, much more work remains – especially in the steps to nominate genes with an allelic series and to test the functional consequences of these alleles on disease-relevant assays.
- Utilize GWAS data and existing annotation pipelines to nominate the most likely causal variant(s) and gene(s) in the region. Although this was done in the IMSGC study, and in general is fairly standard for high-quality genetic studies, it should be possible to expand the approach to include other datasets / features for more accurate annotations. A recent study published in Nature Genetics describes one such pipeline, the Priority index (see here, here).
- Utilize epigenetic and gene expression datasets to nominate most likely pathogenic cell types and tissues. Again, this was done in the IMSGC study, but could be expanded to include other datasets.
- Single cell eQTL / pQTL analysis in pathogenic cells types and tissues. The IMSGC did a nice job on heterogenous tissues (dorsolateral prefrontal cortex, peripheral blood mononuclear cells) and isolated subsets of immune cells (naïve CD4+ T cells, monocytes). However, the analysis overall seems incomplete: it is not clear if co-localization was performed and not clear how the eQTL signals vary across tissues types. Moreover, it is now possible to extend cis-eQTL analysis to single cells via RNA-sequencing (see here), as well as to protein quantitative trait loci (pQTLs). Similarly, it is possible to include gene expression profile not just in resting cells, but also in cells under specific stimulation conditions.
- Refine causal variant / causal gene prioritization. By integrating data from steps 1-3, it should be possible to predict more accurately the most likely causal gene(s) / variant(s) in the region. Indeed, this is an iterative process that should be continuously updated as new information is gleaned.
Most human genetic studies do a good job on these first four steps. However, these steps alone fail to address the 3Ms (mechanism, magnitude, and markers). As a thought experiment, consider the output from the IMSGC study: a long list of new loci; a short list of pathogenic cell types; and refined estimates of heritability explained. But as a drug hunter, how is this actionable? As described below, the next set of steps should begin to address the 3Ms concept by identifying multiple functional alleles that perturb a target in cell types that can be measured as part of a clinical trial.
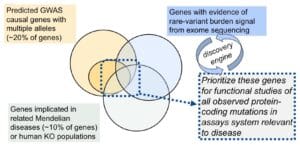
- From this list (see step #4), assess which genes / regions harbor multiple trait-associated alleles, as determined by:
a. Independent alleles from GWAS of the same trait – in the IMSGC paper, they estimate ~20% of MS-associated loci harbor more than 1 risk variant. Whether these variants exert function through the same gene is not clear.
b. Integrate with other datasets to find other trait-associated alleles of larger effect size – which includes:
i. Primary immune deficiency or other Mendelian diseases – a study in rheumatoid arthritis found that ~7% of genes implicated by GWAS were also implicated in Mendelian forms of primary immune deficiency (here).
ii. Human KO from special populations (e.g., consanguineous families, FinnGen) – complete human knockouts provide an estimate of the maximal effect of target perturbation (here, here).
iii. Rare variants from sequencing studies in the same disease – a recent type 2 diabetes sequencing study found “enrichment” in rare-variant burden tests that do not yet reach genome-wide significance (here).
c. Integrate with datasets of quantitative traits – for immune-mediated diseases such as MS, this may include QTLs for blood cell types (here) or protein QTLs from serum (here, here). Mendelian randomization should be performed
d. Integrate with datasets of somatic mutations – while there is much written about somatic mutations in cancer (e.g., COSMIC database), and while these mutations have been used to inform on the 3Ms in rheumatoid arthritis (here), this is an under-utilized source of genetic variation outside of oncology. Clonal hematopoiesis in heart disease represents one interesting example (see NEJM article here), and it is logical that there are other clinical phenotypes where somatic mutations influence disease outcome.
The more alleles that can be identified, the more genetic tools will be available to estimate the range of effect of genotype perturbation on function-phenotype maps. As an example, there are >2000 CFTR mutations that indicate that ~25% restoration of CFTR protein function should be sufficient improve clinical outcome in patients with cystic fibrosis.
Note also that I use the term “trait-associated alleles”, as the alleles do not need to be associated with the exact same phenotype (e.g., risk of MS, risk of cystic fibrosis). While this would be ideal, it is possible to be creative with how phenotypes are considered together along a spectrum of physiology. For immune-mediated diseases such as MS, this range might include risk of infection (complete loss-of-function), protection from autoimmunity (partial loss-of-function), and/or risk of autoinflammation (partial gain-of-function).
- Develop a high-throughput assay system to determine the functional effect of protein-coding mutations that arise from CVAS, RVAS, human knock-outs, Mendelian disease, etc.
a. These assays should be based on functional insights from steps 1-4. In the case of MS, as described in the IMSGC study, there is now strong support for T cells, B cells, natural killer cells, dendritic cells, and microglia, but not for astrocytes or neurons.
b. These assays should be converted to high-throughput assays in order to study a range of alleles, including alleles that have not yet been discovered through genetic association studies (see step #7 below). Examples of such throughout approaches include (and see reviews here, here):
i. Cellular assays (e.g., PPARG as described here, CFTR as reviewed here)
ii. Protein abundance assays (e.g., PTEN as described here)
iii. Enzymatic assays (e.g., PTEN as described here)
iv. Morphological assays (e.g., type 2 diabetes genes as described here)
v. Induced pluripotent stem cells
c. To establish human relevance, these assays should be validated by knocking-out or over-expression genes from step #5. CRISPR and other gene editing technologies make this feasible (see here for a recent announcement from GSK on a new partnership with Jennifer Doudna and Jonathan Weissman). These extreme perturbations should provide an estimation of the assay window.
d. Trait-associated alleles should be introduced into the assay system to estimate the magnitude of effect. This step is critical to provide a function-phenotype relationship, as well as to provide further validation that the assay is disease-relevant. An example is TYK2 – one of the genes implicated in the IMSGC study – where functional studies demonstrated that ~80% loss-of-function was associated with protection from autoimmunity without an increased risk of infection (see here, here, here).
e. Once functionally validated, these assays can be used for a variety of applications: conventional target-based screens against genetic targets; cell-based phenotypic screens around genetic nodes (see here); and functional characterization of additional trait-associated alleles, as they are discovered through population-scale sequencing studies. Indeed, validated functional assays will create a discovery engine to interpret RVAS and to nominate new genes, assays, etc.
- Perform PheWAS for every functionally-validated, trait-associated variant using large-scale biobanks. Population-scale biobanks continue to emerge (e.g., recent announcement of Heredigene, a partnership between Intermountain Healthcare and deCODE) and over time will become fully federated (see efforts such as EMBL-EBI’s ELIXIR). These biobanks can be used for selecting indications for clinical trials, predicting on-target adverse events (here), and selecting biomarkers for clinical trials. The latter can be used as the marker component of the 3M’s framework. For MS-associated alleles that emerge from the IMSGC study, for example, it would be important to query associations with other immune-mediated diseases (indication selection), risk of infection / malignancy (on-target adverse events), and blood traits such as immune cell counts and plasma protein levels (pharmacodynamic biomarkers).
In conclusion, the latest IMSGC study is an elegant scientific study that provides novel insight into the genetic architecture of MS. Nonetheless, the study falls short for what is necessary to posit new therapeutic hypotheses. I propose a 3M framework (mechanism, magnitude, and markers) that geneticists should consider when designing functional follow-up studies. Further, I provide a path forward via an allelic series model, which I believe is applicable across a wide-variety of complex traits. As such studies are pursued, I hope geneticists will move from heel-clicking and wishful-thinking to rigorous functional studies that will deliver novel therapeutic hypotheses.
[A link to a Word and pdf version of this blog can be found here and here. A direct link to the ICDA white paper is here.]