
I admit upfront that this is a self-serving blog, as it promotes a manuscript for which I was directly involved. But I do think it represents a very nice example of the role of human genetics for drug discovery. The concept, which I have discussed before (including my last blog), is that there is a four-step process for progressing from a human genetic discovery to a new target for a drug screen. A slide deck describing these steps and applying them to the findings from the PLoS One manuscript can be found here, which I hope is valuable for those interested in the topic of genetics and drug discovery.
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer. However, the PLoS One study was performed while I was still in academics at BWH/Harvard/Broad.]
Before I provide a summary of the study, I would like to highlight a few recent news stories that highlight that the world thinks this type of information is valuable. First, the state of California is investing US $3-million in a precision medicine project that links genetics and medical records to develop new therapies and diagnostics (here, here). Second, Gonçalo Abecasis and the University of Michigan recently announced Genes for Good, which aims “to work toward cures for generations yet to come by generating and analyzing an enormous database of health and genetic information” (see Slate.com story here). And third, a Mendelian randomization study was published in the NEJM on heart disease and short stature (here). OK, the latter is not exactly related to drug discovery…but it still has to do with the basic principles around human genetics and causality.
Background: What is the strategy to use human genetics for drug discovery? Ideally, genes that harbor a series of disease-associated alleles what a range of functional perturbations – from complete loss-of-function to gain-of-function – are identified. There are a few examples of loss-of-function variants that protect from human disease, and these have proven valuable drug targets (e.g., PCSK9 and coronary artery disease). An open question, however, is how many such genes will be identified from ongoing genetic studies. Further, once identified, it is not clear how these discoveries will lead to new drug targets.
Results: We performed a large-scale genotyping and sequencing study. First, we combined Immunochip dense genotyping (n = 23,092 case/control samples), Exomechip genotyping (n = 18,409 case/control samples) and targeted exon-sequencing (n = 2,236 case/controls samples) to demonstrate that three protein-coding variants in TYK2 (tyrosine kinase 2) independently protect against RA: P1104A (rs34536443, OR = 0.66, P = 2.3×10-21), A928V (rs35018800, OR = 0.53, P = 1.2×10-9), and I684S (rs12720356, OR = 0.86, P = 4.6×10-7). Second, we show that the same three TYK2 variants protect against systemic lupus erythematosus (SLE, P = 6×10-18). Finally, in a phenome-wide association study (PheWAS) assessing >500 phenotypes using electronic medical records (EMR) in >29,000 subjects, we found no convincing evidence for association of P1104A and A928V with complex phenotypes other than autoimmune diseases such as RA, SLE and IBD. Together, our results demonstrate the role of TYK2 in the pathogenesis of RA, SLE and IBD, and provide supporting evidence for TYK2 as a promising drug target for the treatment of autoimmune diseases.
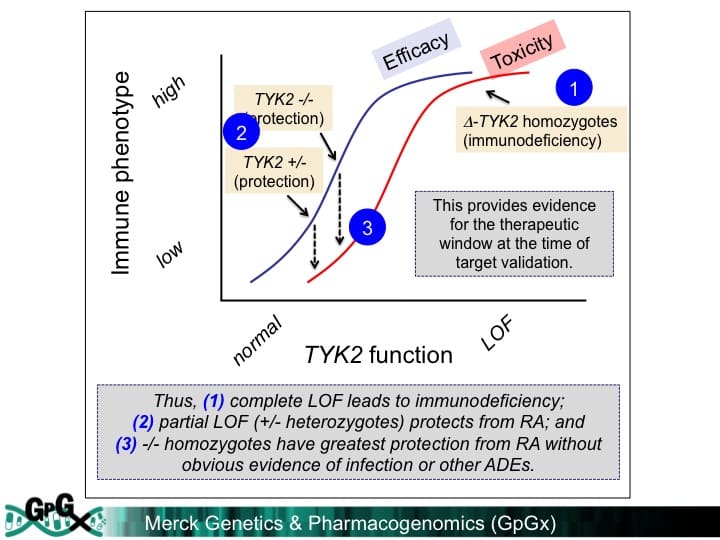
Why this is important: This example adds to the growing literature of LoF variants that protect from human disease. What I really like about the study – admittedly I am completely biased! – is that it provides an example of the “allelic series” model for drug discovery. Complete loss of TYK2 function leads to immunodeficiency. As we demonstrate in this manuscript, partial LoF protects from RA and SLE. But for drug discovery, it is important to estimate the consequences of perturbing a target on both efficacy and safety surrogate endpoints. To estimate therapeutic index, we performed an “adverse event” pleiotropy scan. We used a novel PheWAS approach that links human genetic data to real-world clinical data, much in the way that 23&Me, deCODE, and other “living biobanks” hope utilize genotype + phenotype data for drug discovery. Together, our findings suggest that therapeutic modulation of TYK2 may have a beneficial therapeutic window. That is, if the exact function of the genetic mutations can be deciphered, and if a therapeutic molecule can faithfully recapitulate the genetic perturbation, and if the right amount of TYK2 inhibition can be achieved in humans with disease, then a selective TYK2 inhibitor may be an effective treatment for RA and SLE. Lot’s of “ifs”, but that is why drug discovery is so difficult!