In a previous blog post, I argued that pharmaceutical Research and Development (R&D) is more like poker than chess given the probabilistic nature of developing new medicines. In this blog, I build out this argument further and introduce a new concept, the Critical Value Creation Period (CVCP). The CVCP is the moment in R&D where the probability of success (PoS) for an investigational medicine becoming a new medicine jumps substantially. For most investigational medicines, this period is in early clinical development. By analogy, early development is like the “flop” in Texas hold ’em poker. Stay with me on this one…hopefully this will become clear by the end of this blog post!
Here, I first build out the CVCP argument, which is modeled after an investment thesis proposed by Bain Capital Life Sciences (see here for a presentation by Dr. Adam Koppel at BCLS). Then, I introduce the poker analogy – with focus on the flop. Next, I discuss practical implication of the CVCP model on decision making in biopharma. Finally, I provide two brief case studies that reinforce these concepts. For those not interested in poker, you can skip the second section and focus on the first, third and fourth sections.…
When I last wrote about AI on this blog three years ago, I spoke of it being a tool with the potential to transform scientific discovery, but the application I described was primarily theoretical. For AI to be a meaningful tool in R&D, I argued, we needed better sources of “truth” – better data sets that AI tools could query and learn from over time – and technology capable of integrating multiple steps into a semi-automated system. My message was that AI-enabled drug discovery was coming…someday.
Fast forward to 2025, and that someday is now.
We’ve seen an explosion in the availability and capability of AI tools. Just 10 months after I wrote about the theoretical possibilities of AI in biopharma, OpenAI debuted ChatGPT. Shortly after that, we saw the rollout of Microsoft Copilot and Meta AI. We now have immense computational power at our fingertips, with programs specifically designed to query biological problems. Combined with the ingenuity of skilled scientists, who can define the research problem and generate curated datasets that will enable solutions, AI has become an important and practical tool that is helping researchers accelerate discovery (link to Google DeepMind podcast on this topic here; link to a start-up’s pragmatic journey of AI in drug discovery here).…
Last week I visited the University of Pennsylvania for a fireside chat with Roger Greenberg, a Professor in the Department of Cancer Biology, as part of the Wharton Undergraduate Healthcare Club (WUHC). I shared my personal history and answered questions from an audience primarily consisting of Penn undergraduate students. This inspired me to write down advice that I wish I could have given to my younger self.
This blog is a reflection on the traits of successful people (current tense), with advice I wish I had received as I was navigating the early stages of my career (subjunctive and past tenses). For fun, I used Google’s “Illuminate” app to turn the text below into a 7-minute podcast (podcast audio link here). Oh, the power of Artificial Intelligence!
[Disclaimer: I am a full-time employee of Bristol Myers Squibb. All views expressed here are my own.]
1. Beware of the personal narrative. When established individuals (i.e., old, successful people!) give advice to youth, they often spin a tale that makes sense in retrospect. The narrative may be accurate from an outcomes perspective but is often inaccurate from a journey Further, personal narratives often tell a story that makes the narrator look favorable at the expense of lessons learned for the audience listening.…
Disclaimer: I am a full-time employee of Bristol Myers Squibb.
When I practiced clinical rheumatology, I would often see patients with autoimmune conditions like systemic lupus erythematosus (SLE), systemic sclerosis (scleroderma), rheumatoid arthritis (RA), or myositis. A typical patient journey included an initial sense of relief when a diagnosis was established and a medicine was started. Inevitably, however, there was a sense of dread when a patient would ask: “When am I able to stop taking these strong immunosuppressive medicines?”
I would answer: “Likely never.”
That’s because, historically, there have been no cures for these diseases.
Now, however, I believe there may be an opportunity for new strategies that may deliver transformational outcomes for patients. In a new review paper published in Nature Reviews Drug Discovery (NRDD) (link here), our team at Bristol Myers Squibb (BMS), led by Dr. Francisco Ramírez-Valle, describes a “sequential immunotherapy” strategy that has the goal of achieving durable remissions and even functional cures. I presented an example of this strategy in action for SLE at the Stanford Drug Discovery Symposium. I also discussed the potential for functional cures during a recent BioCentury Show podcast and as part of NRDD’s “An Audience With” series.…
At the beginning of the summer, I had the opportunity to join the team at Royalty Pharma for a great event at MIT (link here). It was an interesting time for me as I was thinking about the new role I was about to take at Bristol Myers Squibb. While I had certainly been “a leader” for several years now, I was pushing myself to think through and question my perspectives on R&D now that I was taking on increasing responsibilities as “the leader” of the research organization.
And so the presentation opportunity with Royalty really pushed me to articulate my views in a way that would hopefully resonate with and inspire others. I titled my presentation “Bullseye.Aim.Fire” and then renamed it “Increasing R&D Productivity to Deliver Transformational Medicines” so the topic would be more obvious. What I’m really sharing in the presentation is my fundamental belief about R&D, linking together several factors that I see as mission critical.
To me, it really all comes down to causal human biology. In order to be successful, we must understand the cause-and-effect relationship between perturbing a particular biological target with a medicine and the outcome that will then impact human physiology.…
For me, the most enjoyable aspect of discovery research is exploring the unknown. It is about having a big idea; believing in that big idea based on a scientific belief framework; coming to a crossroads in the validity of the big idea, which is usually marked by deep uncertainty and skepticism; making a data-driven scientific decision to proceed (or not) to the next inflection point of testing the big idea; and ultimately arriving at a conclusion of whether the big idea is true.
Unfortunately, most of these scientific adventure stories are lost in the way we communicate about science. We tell a story to communicate the final message – we have a new medicine that is effective in treating patients – as that is the cleanest way to communicate to an audience not familiar with the gory details of the discovery. Such retrospective narratives are also the simplest way to communicate the validity of the big idea, not the tortuous and often complicated path to arrive at truth.
But such retrospective narratives don’t capture the immensely personal nature of our research discoveries. Moreover, such retrospective narratives often make the big idea seem preordained or obvious, when the big idea was anything but.…
[ I am an employee of Bristol Myers Squibb. The views expressed here are my own, assuming I am real and not a humanoid. ]
In the original Blade Runner (1982), Harrison Ford’s character, Deckard, implements a fictitious Voight-Kampff test to measure bodily functions such as heart rate and pupillary dilation in response to emotionally provocative questions. The purpose: to establish “truth”, i.e., determine whether an individual is a human or a bioengineered humanoid known as a replicant.
While the Voight-Kampff test was used to establish truth for humans vs replicants, the concept of “truth” is central to neural networks used in machine learning and artificial intelligence (AI). And for AI to be effective in drug discovery and development, it is critical to ask a fundamental question: what is “truth” in drug discovery and development?
INTRODUCTION
I recently read the book Genius Makers by Cade Metz and was reminded of the long history of machine learning, neural networks, and artificial intelligence (AI). This is a field more than 60 years in the making, with slow growth for the first 50 years – AI was founded as an academic discipline in 1956 – and exponential growth in the last 10. The original mathematical framework of neural networks was created in the 50’s (perceptron), 60’s and 70’s (backpropagation), but went largely unappreciated outside of academics, as the practical applications were few and far between.…
[Disclaimers: I am an employee of Bristol Myers Squibb. The views expressed here are my own. Also, I am not a particularly good poker or chess player. It is one reason I am a popular invited guest to poker nights with friends.]
I posted on poll on Twitter to ask the question is drug discovery more like poker or chess. There were over 300 responses, with the results split nearly equally (54% poker, 46% chess).
My answer to the question, “Is drug discovery more like poker or chess?”, derives from the following truths:
Poker is a game of skill and chance, where critical information about how to win is hidden. In poker, one has to make probabilistic decisions with incomplete information.
Chess is a game of pure skill, where all information is available and – for the best players – decisions are deterministic. Unlike poker, chess contains no hidden information and very little luck.
Thus, my “answer” to the question is drug discovery is more like poker than chess – largely because of available information (poker = incomplete, chess = complete) and the importance on probabilistic (poker) vs deterministic (chess) decision-making. Here is more context.
Thinking in bets
I recently read the book “Thinking in Bets” by poker champion Annie Duke (@AnnieDuke).…
[Disclaimer: I am an employee of Bristol Myers Squibb. The views expressed here are my own.]
One of my favorite questions to ask is: “What captures your imagination?” At a recent family dinner, responses were varied but encouraging for the next generation: black swan events, comparative anatomy & human physiology, space exploration & intelligent life beyond our planet, and more. My response was programmable therapeutics, a topic which I have blogged about in the past.
In this blog I define programmable therapeutics and provide a few recent examples (severe combined immune deficiency and mRNA vaccines). As you will see, programmable therapeutics is more than pure imagination – we are seeing this new concept evolve before our very eyes.
What is the concept of programmable therapeutics?
While there are different definitions of the concept of programmable therapeutics (see a16z talk; programmable cells; synthetic biology; CRISPR base editing), my definition of programmable therapeutics relates to a platform with modular components that can shorten the time from new target to drug candidate and ultimately regulatory trials that can lead to an approved medicine.
For most drug development programs, the identification of a drug target represents the start of a long journey that is highly artisanal.…
(Disclaimer: I am an employee of BMS. The opinions expressed here are my own.)
Do you ever wonder what leadership skills are required to advance an idea from concept to the precipice of regulatory approval? In this guest blog, Dr. Kristen Hege, senior vice president of Early Clinical Development, Hematology/Oncology & Cell Therapy at Bristol Myers Squibb (BMS), describes a harrowing tale hiking in the Sierra Mountains and what it taught her about leadership.
For background, Kristen has been working on cell therapies for more than two decades (link to BMS profile here, Nature Medicine interview here). As a post-doc and, eventually, head of clinical development at a now defunct Bay Area biotech company in the 1990’s, she did research on genetically engineered T cells and hematopoietic stem cells to redirect the immune system to specifically target and kill cancer and HIV-infected cells. More recently, Kristen was part of a team who published a pivotal study of a BCMA CAR-T therapy, ide-cel (or bb2121) in patients with relapsed and refractory myeloma (link to NEJM article here).
But here you will see another side of Kristen, who is widely revered among her biopharma peers as an adventurer par excellence.…
And now for something completely different on my plengegen.com blog. This blog was written in partnership with a number of scientists and clinicians in my local community and serves as rationale for comprehensive viral testing program in K-12 schools. The document (pdf link here) was circulated to members of our community and discussed as part of a town webinar last week (link here).
Executive summary:
There is much debate about the value of viral testing program for K-12 schools. As described here, we support a comprehensive viral testing plan that returns results in less than 24 hours as part of an overall risk reduction strategy. Testing should be prioritized as follows: (1) baseline “time zero” at the start of school of all students, teachers and staff; (2) symptomatic testing of all students, teachers and staff; (3) at least weekly surveillance testing of all teachers and staff; (4) at least weekly surveillance testing of older students; and (5) at least weekly surveillance testing of younger students. While symptomatic testing is available through local hospitals, clinics and testing centers, baseline time zero testing and surveillance testing of asymptomatic individuals is not. Thus, additional investment is required to support a comprehensive viral testing program in K-12 schools.…
[I am an employee of BMS. The views expressed here are my own.]
The blog is long, so I will start with an executive summary. (You can download a pdf copy of the blog here.) Pharmacologic intervention has the opportunity to impact disease progression in the SARS-CoV-2 / COVID-19 crisis. Repurposing of approved therapies is the fastest way to impact patients today, as these medicines have regulatory approval to enable investigator-initiated trials and have a manufacturing process to ensure drug supply. Here, I focus on a specific clinical inflection point in COVID-19 disease progression – hospitalized patients early in their disease course and with signs of a maladaptive immune response, with the intervention intended to prevent disease progression and admission to the ICU. Based on an understanding of disease biology today – which is still quite limited – this clinical inflection point is due to a “maladaptive immune response” seen early in the disease course in patients who later progress to critical illness. Rigorous clinical trials are required to test therapeutic hypotheses related to repurposed therapies, which need to be done in a clinical setting caring for extremely sick patients. Finally, I describe additional research that is required to understand the biology of SARS-CoV-2 / COVID-19, and how such research (e.g.,…
[I am an employee of Bristol-Myers Squibb. The views expressed here are my own.]
One of my predictions for the next decade – the “clear view” decade – is that we will have the ability to click on any gene in the human genome to generate function-phenotype maps. These maps should enable drug discovery by informing on mechanism, magnitude and markers of target perturbation. In particular, I have championed an “allelic series” model, whereby genes with a series of alleles are used to derived genetic dose-response curves (see here, here).
During a recent presentation to my former colleagues at the Division of Genetics at Brigham & Women’s Hospital (BHW, slides here), I discussed important assumptions underlying this model:
Large-scale sequence data will identify a range of protein-coding variants associated with traits of medical interest that are suitable surrogates for drug discovery (allelic series architecture assumption).
It will be possible to use high-throughput functional assays to interrogate the impact of trait-associated variants on cell physiology for the majority of genes in the genome (functional readout assumption).
Large-scale biobanks will emerge to enable testing of these same trait-associated variants for pleiotropic effects across a wide-variety of clinical phenotypes in the real world (PheWAS assumption).
[I am an employee of Celgene. The views expressed here are my own.]
In the Wizard of Oz, Dorothy clicks her heels and hopes for re-entry from her dream world by repeating, “There’s no place like home…there’s no place like home…” I often feel that many in the genetics community look at their human genetics data with the same youthful optimism as Dorothy – clicking their genetic heels and wishing “my genetic discovery will become a drug…my genetic discovery will become a drug…” But without rigor and discipline, such heel-clicking won’t overcome many of the challenges that face drug hunters along the tortuous journey from a genetic idea to a new medicine.
In this blog, I discuss a recent study on the genetics of multiple sclerosis (MS) published in Science (see here). This is a beautiful study that substantially advances the genetic landscape of patients with a devastating disease. However, the study falls short in terms of the application of human genetics to drug discovery. To chart a course for the future, I introduce the concept of mechanism, magnitude and markers (oh my!), which I refer to as the three M’s. …
[Disclaimer: I am an employee of Celgene. The views expressed here are my own.]
Human genetics offers the potential to identify drug targets and to inform decision-making on the journey to an approved drug. A recent study by Ference et al in the New England Journal of Medicine (NEJM) provides an example of human genetics in action. While most of the study focuses on Mendelian randomization to establish a relation among ACLY genetic variation, LDL cholesterol levels, and cardiovascular events, in this blog I focus on a topic highlighted in the companion NEJM editorial: human genetics to predict on-target adverse drug events (see NEJM editorial here).
First, what is the framework for the application of human genetics to predict on-target adverse drug events (ADEs)? Briefly, human genetics can predict on-target toxicity if the following criteria are met: (1) unambiguous association of genetic variant to a clinical phenotype that is a surrogate for drug efficacy and toxicity; (2) unambiguous relationship between disease-associated variant and implicated gene that is the target of the therapeutic intervention; (3) quantitative assessment of gene function and clinical phenotypes of efficacy and toxicity to estimate a “genotype-phenotype dose-response” relationship; and (4) confidence that the therapeutic intervention mimics the mechanism of action of the disease-associated variant.…
[I am an employee of Celgene. All views expressed here are my own.]
At the 2018 Annual Atlas Ventures Retreat (AVR), I participated in a panel on Digital Health (along with David Schenkhein, John Reed, Scott Brun). The panel discussion was led by Michael Ringel, who also provide an excellent introduction to Digital Health (his slides here). While there are many aspects to digital health, we focused on the application to drug discovery and development. In this blog, the main point I want to emphasize is that I believe that the digital health tipping point will occur when products that benefit patients (e.g., therapeutics) facilitate the integration of digital health initiatives that currently reside in silos.
What is digital health in relation to drug discovery & development? There are many different definitions with many different components, and this, in essence, is part of the challenge (see Figure below). In early discovery biology, digital health represents various data types (e.g., human genetics, ‘omics data, cell models) and analytical methods (e.g., simple regression, machine learning, artificial intelligence). In late discovery biology, digital health includes sophisticated analytical methods for in silico drug design and organoid models to recapitulate the human system for pre-clinical testing.…
[I am an employee of Celgene. All opinions expressed here are my own.]
A meeting was recently convened to discuss a roadmap for understanding the genetics of common diseases (search Twitter for #cdcoxf18). I presented my vision of a genetics dose-response portal (slides here; link to related 2018 ASHG talk here). The organizers (@RachelGLiao, @markmccarthyoxf, @ceclindgren, Rory Collins [Oxford], Judy Cho [New York], @NancyGenetics, @dalygene, @eric_lander) asked participants to share their vision. I thought I would blog about my mine.
You’ll notice my vision is ambitious. Nonetheless, I believe these objectives are feasible to accomplish within a 3-year (Phase 1) and 7-year (Phase 2) time frame. Phase 1 would start immediately and would guide projects for Phase 2. In reality, many aspects of Phase 1 are already underway today (e.g., GWAS catalogue at EBI; Global Alliance for Genomics and Health [GA4GH] data sharing methods). Phase 2 consists of two parts: federation of global biobanks and experimental validation of variants, genes and pathways. Some components of Phase 2 could start today (e.g., exome sequencing in >100,000 cases selected from existing case-control cohorts and biobanks; human knockout project). As with Phase 1, many components of Phase 2 are already underway (federation of existing biobanks [e.g.,…
[Disclaimer: I am an employee of Celgene. The views reported here are my own.]
I presented at the PharmacoGenomics Research Network (PGRN) portion of the 2018 ASHG meeting (link to my slides here). A major theme from my talk was that precision medicine holds promise for advancing novel therapies, but that implementation of pharmacogenomics (PGx) will happen by design not by accident. Here is what I mean – and why our health care systems need to build for this future state today.
PGx by design – PGx by design starts at the very beginning of the drug discovery journey, when the choice is made to develop a therapeutic molecule against a target or a pathway. A precision medicine hypothesis is carried forward into the design of a therapeutic molecule (“matching modality with mechanism”), pre-clinical biomarkers to measure pharmacodynamic responses, and early proof-of-concept clinical studies in defined patient subsets. Late-stage clinical development is performed in these patient subsets, and regulatory approval is obtained with a label that defines this patient subset. Health care systems will essentially be required to incorporate precision medicine into patient care.
There are emerging examples of PGx by design. Indeed, there are an increasing number of FDA approvals that fit with the PGx by design model (see figure below).…
[I am an employee of Celgene. All views expressed here are my own.]
What is the clinical significance of residing within the tail of a distribution for disease risk? A new study published in Nature Genetics uses a composite polygenic score to measure extremes of genetic risk(see original article here). The authors make the bold statement: “it is time to contemplate the inclusion of polygenic risk prediction in clinical care”. In this plengegen.com blog, I briefly review the paper, frame the impact of the study in terms of “long tails”, and propose how genetic tails may be used as part of a healthcare system reimagined.
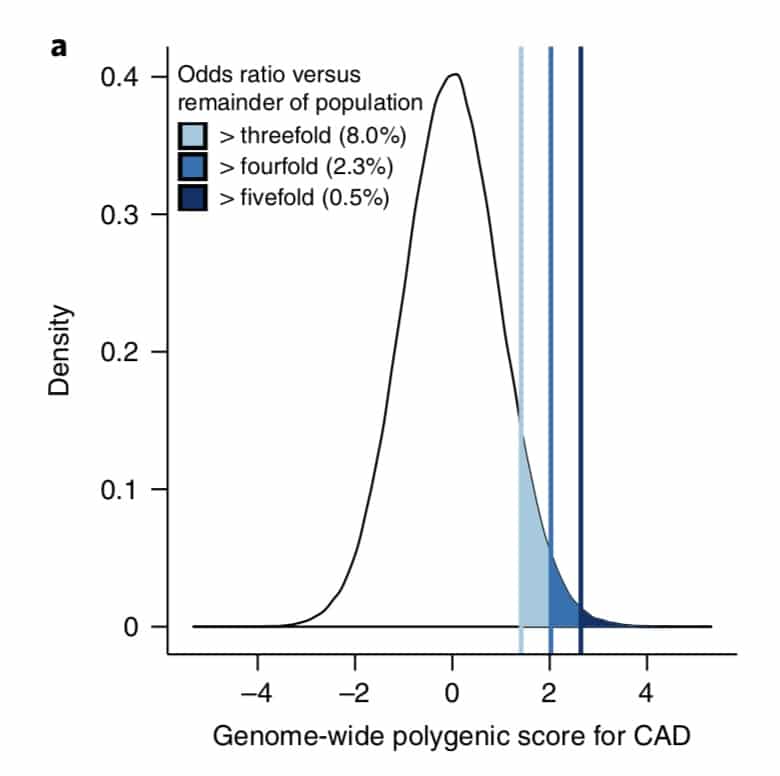
The premise of the paper is that a genome-wide polygenic score (GPS) – a composite genetic test that includes thousands and sometimes millions of genetic variants – can identify a small number of individuals from the general population that have an elevated risk. The study applies polygenic risk scores to five common diseases but spends most attention to coronary artery disease (CAD). For each disease, the increase in risk is approximately 3- to 5-fold higher among individuals at the extreme of the polygenic tail compared to those in the general population – see Figure 2a (and below) for CAD, where ~8% of the general population is at a 3-fold increase in risk based on a polygenic risk score.…
[Disclaimer: I am an employee of Celgene. The views reported here are my own.]
I recently participated in a Harvard Medical School Executive Education course on human genetics and drug discovery (link here, slides here and here). My presentation concluded with a short discussion on emerging resources such as Phenome-Wide Association Studies (PheWAS) to predict adverse drug events and guide indication selection, and protein quantitative trait loci (pQTLs) for Mendelian randomization. In this blog, I highlight briefly our recent Nature publication on pQTLs, “Genomic atlas of the human plasma proteome” (here), which represents a new public resource for drug discovery.
Human genetic targets are endowed with favorable properties, one of which is the ability to use genetic tools for nature’s randomized control trial. Central to this concept is Mendelian randomization, a method that uses human genetic variants as an instrument to examine the causal effect of a modifiable exposure (e.g., protein biomarker) on disease in observational studies (reviewed here and recent Nature Reviews Geneticshere).
Proteins provide an ideal paradigm for Mendelian randomization analysis for drug discovery, as proteins are under proximal genetic control and represent the targets of most approved drugs.…




















{kind=link}