
I have many fears, both professional and personal. When I decided to leave academics for a job in industry in 2013, my biggest fear about making the transition was scientific. In my mind, I had a model of how human genetics might transform drug discovery and development. There were anecdotes (e.g., PCSK9 inhibitors) and a few systematic studies in specific diseases (e.g., genetics of rheumatoid arthritis), but there were many holes to the model. Over the last couple of years, additional anecdotes and systematic analyses have emerged (e.g., Matt Nelson, et al. Nature Genetics), which helps to soothe my fears…but I still have concerns.
[Disclaimer: I am a Merck/MSD employee. The opinions I am expressing are my own and do not necessarily represent the position of my employer.]
As I have blogged about previously, I see two primary routes to go from human genetics to new drug discovery programs (see here, here). The first requires that there are genes with a series of disease-associated alleles with a range of biological effects, ideally from gain- to loss-of-function (allelic series model). The second requires disease-associated genes to aggregate within specific biological pathways, which can then be turned into assays for disease-relevant pathway-based screens such as phenotypic screens. In both, there is an underlying assumption, which is now well validated, that most complex traits are highly polygenic.
In truth, there are uncertainties with both models – and this is at the heart of my professional fear. How many genes will really have a series of alleles for which biological function can be ascertained? Once identified, can those genetic targets be drugged? Can genetic pathways be turned into robust assays relevant for early drug discovery programs? This keeps me up at night.
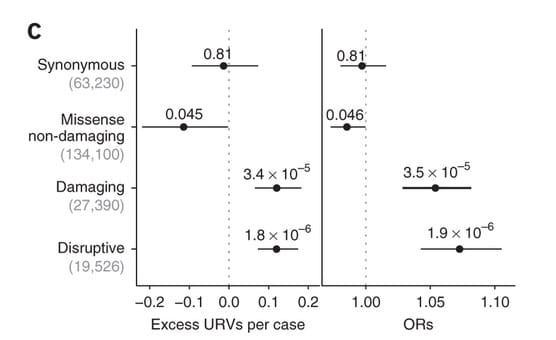
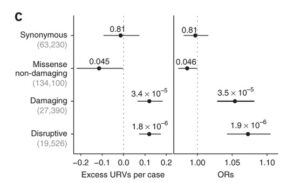
A new study in Nature Neuroscience by Steve McCarroll and colleagues provides me with some comfort, but it also raises important questions about polygenic models for drug discovery. The study performed whole exome sequencing (WES) in 4,946 affected individuals with schizophrenia and 6,242 unaffected controls from Sweden. They filtered variants to identify a set of “ultra-rare variants” (or URVs), which they defined as variants seen only in a single individual, regardless of case-control status. Using in silico tools, they classified URVs based on putative function: disruptive, damaging, missense non-damaging, and synonymous. As shown in Figure 1C (and below), they observed significant case-control differences in the rates of disruptive URVs (n=19,526 variants, P = 1.8 × 10−6) and damaging URVs (n=27,390 variants, P = 3.4 × 10−5).
{kind=link}
Next, they performed a variety of gene set enrichment tests and found an excess of URVs in genes previously implicated in genetic studies schizophrenia and related neurological disorder, including (a) X-linked intellectual disability genes (OR = 1.88; P = 9.5 × 10−4), (b) developmental disorder genes (OR = 1.67; P = 1.6 × 10−4), (c) genes overlapping de novo deletions previously found in individuals with schizophrenia (OR = 1.34; P = 0.0052), and (d) GWAS of schizophrenia (OR = 1.37; P = 0.027), amongst other gene sets. These findings provide empiric evidence for the allelic series model. I note, however, that the overlap with GWAS hits in particular is at only a suggestive level of statistical significance given the number of hypotheses tested.
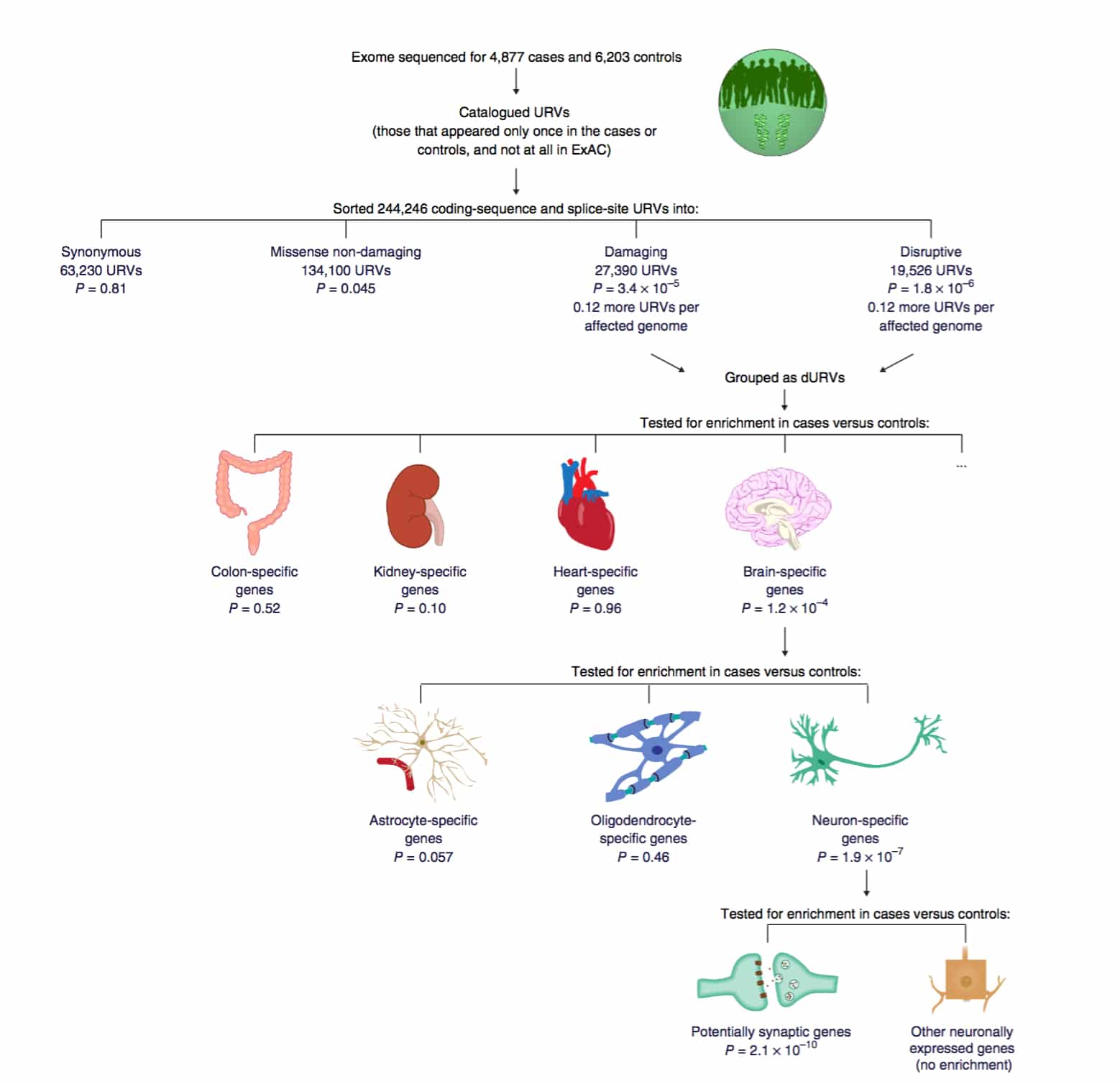
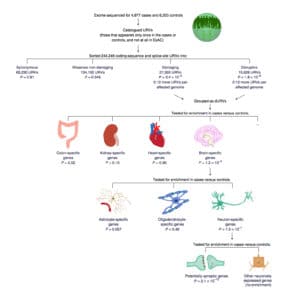
As summarized nicely in Figure 6, the study also found enrichment of URVs in genes that are specifically expressed in neurons, rather than in other CNS cell types (P = 2.1 × 10−10). Such pathway analysis provides empiric support for the genetic pathway model.
{kind=link}
But here is the rub. There are two challenges to use the output of this elegant genetic study to guide drug discovery:
(1) The study was not able to resolve most of the URV signal to individual genes. Through gene burden tests, no single gene emerged as surpassing a study-wide level of significance (Supplementary Figure 4). There was, however, suggestive evidence for a few genes previously implicated in rare variant studies in schizophrenia (e.g., TAF13, SETD1A, NRXN1, see Supplementary Table 3). For example, a previous study identified rare loss-of-function variants in SETD1A as associated with increased risk for schizophrenia and learning disabilities at a genome-wide level of significance (here). Thus, for the allelic series model to successfully identify new disease genes – and therefore new drug targets – many more individuals need to be sequenced.
How many more? The manuscript only states “much larger cohorts will be needed”. No modeling is provided to estimate cohort size. The study referenced Zuk et al PNAS 2014, which provided estimates that at least “at least 25,000 cases, together with a substantial replication set” is required for a well-powered rare-variant association study.
(2) While polygenic models identify genetic pathways, it is a challenge to make this actionable for drug discovery. Through sophisticated pathway analyses, the study implicated “neuron-specific genes, but not in other brain cell type specific genes”. The authors hypothesize that synaptic genes in particular are relevant to the pathogenesis of schizophrenia. The challenge is to turn that gene set into a robust assay for a phenotypic screen, or into discrete biology to select compelling targets. This is not an easy task. Indeed, a recent Nature Reviews Drug Discovery article by Horvath and colleagues proposed a set of principles to facilitate the definition and development of disease-relevant assays.
Finally, I should briefly point out two companion papers (here, here). The first, by Pamela Sklar and colleagues, used gene expression data from brain tissue (dorsolateral prefrontal cortex, to be exact) from people with schizophrenia (N = 258) and control subjects (N = 279) to create co-expression networks. Then, they altered expression of specific genes in zebrafish and in human neural progenitor cells to gain insight into human physiology. The second, by Ben Neale and colleagues, demonstrated that disruptive/damaging URVs in highly constrained genes influence the determinants of years of education in the general population.
OK – so those are my professional fears to leverage human genetics to drive drug R&D. As for my personal fears…that is a conversation for another time!