

[I am an employee of Bristol-Myers Squibb. The views expressed here are my own.]
One of my predictions for the next decade – the “clear view” decade – is that we will have the ability to click on any gene in the human genome to generate function-phenotype maps. These maps should enable drug discovery by informing on mechanism, magnitude and markers of target perturbation. In particular, I have championed an “allelic series” model, whereby genes with a series of alleles are used to derived genetic dose-response curves (see here, here).
During a recent presentation to my former colleagues at the Division of Genetics at Brigham & Women’s Hospital (BHW, slides here), I discussed important assumptions underlying this model:
- Large-scale sequence data will identify a range of protein-coding variants associated with traits of medical interest that are suitable surrogates for drug discovery (allelic series architecture assumption).
- It will be possible to use high-throughput functional assays to interrogate the impact of trait-associated variants on cell physiology for the majority of genes in the genome (functional readout assumption).
- Large-scale biobanks will emerge to enable testing of these same trait-associated variants for pleiotropic effects across a wide-variety of clinical phenotypes in the real world (PheWAS assumption).
In this blog, I provide background information and recent examples from the published literature to provide support that these underlying assumptions will be proven true. (1) To address the first assumption, I review data on germline genetic variation and highlight two recent Nature papers on somatic mutations in ulcerative colitis (here, here; see also bioRxiv post here). These two papers provide an interesting twist on the allelic series model: ability to use somatic mutations to understand gene function. (2) To address the second assumption, I review the concept of saturation mutagenesis (tweet here), highlighting a 2018 Nature study on BRCA1 (here), as well as discuss a recent Nature Genetics from Charles Fulco, Jesse Engreitz, and colleagues on a high-throughput experimental approach to perturb enhancers (here). (3) To address the third assumption, I highlight a recent high-throughput phenotyping method, PheCAP, to extract information from electronic medical records (here), as well a study from the UK Biobank, which describes a disease-agnostic approach to cluster the genetic risk across nearly 20,000 disease classification codes (here).
Finally, I conclude the blog with an example of how this might be done for genes within the type 1 interferon pathway. I highlight this as something feasible within the International Common Disease Alliance (ICDA), Open Targets, Lupus Research Alliance, or any individual academic / industry lab. Indeed, I hope as a broad community we are able to rigorously test the allelic series model over the next 3-5 years.
1.What is the probability of the first assumption being true?
1A. Background
In my BWH presentation, I emphasized four lines of evidence to support the hypothesis that there will be a large number of genes that will fit with the allelic series model (see Figure below, slides #35-29 in the presentation).

In my opinion, based on the evidence above, there will be a sizeable number of genes – probably a majority of genes in the genome – that harbor multiple trait-associated alleles. What is not clear is how many trait-associated alleles any given gene will harbor, nor is it clear if there will be a sufficient number of alleles within these genes to generate genetic dose-response curves.
I think the tipping point for the allelic series model will be the availability of large-scale exome sequencing data in case-control cohorts. Very few sufficiently powered studies have been conducted to date. Population genetics theory predicts that at least 25,000 cases (and likely >100,000 cases) and an equal or greater number of controls are required for well-powered rare-variant association studies (here). Indeed, as such studies are published (e.g., type 2 diabetes [T2D] Nature paper here) or presented (e.g., schizophrenia ASHG plenary presentation here), genes with an allelic series are starting to be identified. If these studies find a large number of protein-coding mutations associated with traits of interest, then the bottleneck will quickly become functional interrogation (see next section).
However, the exact number of predicted rare variants per gene is heavily influenced by the underlying disease and gene function, which in turned is influenced by evolutionary selection. For some diseases (e.g., type 2 diabetes, inflammatory bowel disease), where selective pressure is minimal, it seems likely that disease-associated genes will harbor a large number of low-frequency disease-associated variants suitable for PheWAS in independent populations. In the Flannick et al Nature study (2019), SLC30A8 was found to harbor more than 100 variants, at least 30 of which were found to protect from T2D upon independent replication. In contrast, other diseases are likely subject to purifying selection (e.g., schizophrenia), thereby preventing high-risk alleles from reaching even modest allele frequencies (here). Empirical data support this theoretical prediction: most of the rare variants in schizophrenia fall into the “ultra-rare” category (here).
1B New published examples
But what I have not emphasized much is the contribution from somatic mutations to the allelic series model. That is, somatic mutations also serve as a source of genetic diversity to inform on function-phenotype relationships for genetic dose-response curves. While most scientists are quite familiar with the role of somatic mutations in cancer, there is an emerging literature on somatic mutations in heart disease (here), skin (here) and esophagus (here).
In two recent papers published in Nature, the literature on somatic mutations is expanded to include ulcerative colitis, an inflammatory disease of the colon (here, here; also bioRxiv preprint from Carl Anderson’s lab here). Both Nature studies show that the inflamed intestine undergoes widespread re-modelling by pervasive clones, many of which are positively selected by acquiring loss-of-function (LoF) mutations in genes from the IL17-NFkB signaling pathway. The mutated genes include NFKBIZ and TRAF3IP2, both of which are also implicated in GWAS of inflammatory bowel disease (IBD). Under normal conditions, IL17A induces apoptotic cell death. However, in UC, somatic LoF mutations in the IL17A-NFkB signaling pathway confer a local survival advantage under conditions of chronic inflammation.
One intriguing aspect of the Nanki et al study is that they used gene editing in human organoids to characterize the biological effects of mutated genes. Using a CRISPR-based approach, they were able to identify genes that, when knocked-out, confer a survival advantage during IL17A-mediated positive selection. While not performed by Nanki and colleagues, it should be possible to use this same organoid assay system to characterize a range of protein-coding mutations within IL17-NFkB signaling genes. For example, it should be possible to perform saturation mutagenesis of all protein-coding mutations in NFKBIZ and TRAF3IP2 and determine whether the mutations impair IL17-NFkB signaling. As germline exome sequencing data becomes available in UC case-control samples, it should be possible to determine which mutations are functional in the organoid assay.
Whether somatic mutations add substantially to function-phenotype maps for drug discovery outside of oncology is not yet clear. It seems that any naturally occurring variants will add to knowledge about human biology. My guess is that somatic mutations will be particularly useful in understanding function for those genes that also harbor trait-associated germline variants (e.g., NFKBIZ and TRAF3IP2). It will take creative thinking, however, to incorporate biological information derived from somatic mutations into genetic dose-response curves derived mainly from germline trait-associated variants.
1C. Interim assessment of allelic series architecture assumption
Based on available data, it is highly likely that this assumption will be proven true based on germline trait-associated variants alone. For these genes, somatic variants will likely also contribute useful function-phenotype information, although creative thinking will be required to harmonized functional and phenotype data with that derived from germline variants. What is less clear is whether the number of trait-associated germline alleles will be modest in size (i.e., <10 per gene) or sufficiently large (e.g., >30 per gene) to generate function-phenotype maps for any given gene. Population genetics theory and available sequencing data suggest that the exact number will vary by indication and gene function.
2. What is the probability of the second assumption being true?
2A Background
An important unanswered question is not if genes will harbor multiple trait-associated alleles but will there be a sufficient number of trait-associated alleles with a range of functional effects (from loss-of-function to gain-of-function) to generate genetic dose-response curves. As described above, I think the tipping point for function-phenotype maps will be the availability of large-scale case-control exome sequencing data. As data are generated, population genetics predicts that, for certain diseases not under strong purifying selection (e.g., T2D, IBD), there will be a large number of protein-coding mutations of uncertain functional significance. Add to this the number of mutations identified via Mendelian sequencing studies – and potentially somatic sequencing studies – and the number of potential pathogenic mutations per gene becomes quite large. To study function of these mutations – especially at scale across the genome – new approaches to functional characterization are required.
One idea is to perform saturation mutagenesis in advance of large-scale sequencing studies. The idea is that every possible mutation is introduced into a high-throughput assay system that can provide a quantitative (or qualitative) readout of gene function. The value proposition of this pre-emptive approach is that it models all possible mutations, not just those trait-associated variants discovered to date. As new mutations are identified in sequencing studies, it should be possible to quickly look-up the functional impact of these mutations. Incorporating functional data should be quite useful in determining, in real-time, if the mutation is likely to be trait-associated or not.
Saturation mutagenesis is increasingly possible (tweet with references here). One example, published by Jay Shendure and colleagues in 2018, described saturation genome editing (SGE) of all possible single-nucleotide variants (SNVs) in 13 exons of BRCA1 (see here). The study used a haploid cell line (HAP1) whose survival was dependent on critical genes such as BRCA1. If genome-editing of a specific amino acid leads to a non-functioning BRCA1 protein, then the cell will die; if the edited amino acid has no functional impact on BRCA1, then the cell will live. Via SGE and high-throughput sequencing the study investigated nearly 4,000 SNVs. As described below, it should be possible to use a similar approach for genes that harbor a series of trait-associated alleles.
2B New published example
A recent Nature Genetics study by Jesse Engreitz and colleagues describes a high-throughput method, CRISPRi-FlowFISH, that incorporates data from CRISPR interference, RNA fluorescence in situ hybridization (FISH) and flow cytometry to derive insight on non-coding elements across the genome (article here, tweetorial here). While not related to protein-coding variants that I believe will be critical in generating genetic dose-response curves, the study is an excellent example of creative engineering to use high-throughput biology to resolve important questions. As described in Figure 1 of the study: “Cells expressing KRAB-dCas9 are infected with a pool of gRNAs targeting DHS elements near a gene of interest, labeled using RNA FISH against that gene and sorted into bins of fluorescence signal by FACS. The quantitative effect of each gRNA on the expression of the gene is determined by sequencing the gRNAs within each bin.” Of 4,000 perturbed distal element-gene (DE-G) pairs, approximately 4% led to significant changes in gene expression in K562 human erythroleukemia cells. Using these data, the authors developed a new model, termed the “activity-by-contact (ABC) model”, to predict enhancer–gene connections. They then applied the ABC model to predict 997 measured DE-G pairs in five additional human and mouse cell types, as well as to regulatory regions implicated by GWAS. The authors conclude that these observations suggest “a systematic approach to decoding of transcriptional regulatory networks and to interpret the functions of noncoding genetic variants that influence human traits.”
2C. Interim assessment of functional readout assumption
With SGE and clever assay engineering, it is highly likely that this assumption will be proven true. That is, it should be possible to develop high-throughput assays for saturation mutagenesis of individual genes, as described for protein-coding variants in BRCA1 and regulatory regions using CRISPRi-FlowFISH. As per section 1, however, what is not clear is whether there will be a sufficient number of trait-associated alleles to make this pre-emptive investment worthwhile. A reasonable investment at this time would be to nominate genes that seem likely to harbor multiple trait-associated alleles (see figure below) and build high-throughput assays to study the effect of all protein-coding mutations. At the very end of this blog I provide an example of one such approach for the type 1 interferon pathway – as well as emphasize an important consider when designing these high-throughput functional assays.
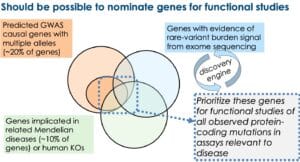
3.What is the probability of the third assumption being true?
3A. Background
Of the three assumptions, I think the ability to perform phenotyping at scale using real-world data is most likely to be proven true. Indeed, there is now a Wikipedia page for biobanks! A simple survey of biobanks across the world reinforces this position (links here, here, here). The UK Biobank (UKBB) is perhaps the shining example of what a biobank can do, but there are many others, too (e.g., FinnGen, All-of-Us, deCODE, Million Veterans Project, Biobank Japan, China Kadoorie Biobank).
A limitation is the ability to harmonize clinical and genetic data across the multitude of biobanks. Take “rheumatoid arthritis” (RA) or “inflammatory bowel disease” (IBD) as what may appear as simple examples at first blush (embedded genomics links here). Some biobanks rely on diagnostic codes to define which individuals suffer from these diseases. Others use patient self-report. And others build algorithms that incorporate structured data (e.g., diagnostic codes, laboratory values, medication history) and unstructured data (e.g., physician notes). All are valid approaches, although the sensitivity, specificity, and positive predictive value will vary.
Moreover, data from biobanks are incompletely harmonized with data from the published literature. (e.g., GWAS Catalogue). Companies such as Genomics plc and public consortium such as Open Targets have done a good job of harmonizing publicly available data. Still, few companies or biobanks have yet tackled the onerous job of harmonizing published data with biobank data, and to federate data across multiple biobanks.
2B New published examples
A recent study published in Nature Protocols (here) describes a high-throughput semi-supervised phenotyping pipeline that begins with EMR data and outputs a phenotype algorithm, the probability of the phenotype for all patients, and a phenotype classification (yes or no). (Disclosure: I am a co-author.) This is a project conceived of nearly ten years ago, as several of us, including senior authors Kat Liao, Tianxi Cai, and Zak Kohane, were reviewing EMR clinical data to establish algorithms for RA and IBD. The process was laborious: we manually reviewed charts to establish a gold standard data set on which to train and test algorithms using structured and unstructured data. While we were excited about the prospects of using EMR data to define a wide-range of phenotypes, we were daunted by the prospects of what it would take to do this efficiently and accurately for more than a few phenotypes. From this problem statement emerged PheCAP. The main breakthrough of PheCAP is the ability to standardized steps in creating EMR algorithms, which in turn improves efficiency, facilitates data checks as well as replication across institutions (see Figure below).
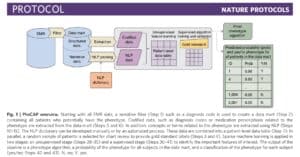
An important assumption of PheCAP is that conventional definitions of disease represent the right classification scheme. For example, there are distinct diagnostic criteria for immune-mediated diseases such as lupus, rheumatoid arthritis, inflammatory bowel disease, and psoriasis, suggesting that these represent distinct entities. In the real-world clinical setting, there is substantial overlap among these diseases: an overlap between RA and lupus is referred to as “rhupus” and patients with psoriasis or IBD often suffer from an inflammatory arthritis with features similar to RA. Thus, while conventional definitions are useful in clinical practice today, these definitions may be too simplistic in terms of capturing the underlying biological processes driving disease.
A second study, published by Gil McVean and colleagues in Nature Genetics, implements a novel statistical approach, TreeWAS, for analysis of genotype-phenotype relationships in the UK Biobank (here). TreeWAS refers to a Bayesian approach for mapping genetic risk across disease classification codes within a hierarchical ontology. The study used TreeWAS to identify groups of variants that have similar impact across diseases. These groups of variants were used as genetic instruments to establish causal relationships among phenotypes (e.g., risk of hypertension, diabetes, kidney disease).
The main point to emphasize here is not the details of either study, but the fact that new methodologies are being developed and applied to large-scale biobank data (PheCAP to define phenotypes at scale, TreeWAS to analyze clusters of variants across clinical ontologies). These methodologies, along with many others, will continue to mature in the coming years. It is also worth noting that real-world data are increasingly used in drug development (WSJ article here), which should create a virtuous cycle where biobanks are used to establish therapeutic hypotheses early in discovery and then used to test these hypotheses years later in registration-scale clinical trials.
3C. Interim assessment of PheWAS assumption
There has been continued growth of biobanks in which genetic data are linked to clinical data. New methodologies are being developed and will almost certainly continue to be developed and deployed within these biobanks, as shown here for PheCAP and TreeWAS. The main limitations are the harmonization of phenotypes and the ability to federate results across biobanks to establish extremely large sample sizes. Given the burning platform to accomplish harmonization of real-world data, and the potential of a virtuous cycle from discovery through registrational clinical trials, I believe it is highly likely that this assumption will be proven true within the decade.
4. What is an example?
In this final section, I provide an example of how all of these components can be brought together to test the allelic series model. Such use cases are very important to rigorously test the assumptions described in this blog. I believe that organizations such as International Common Disease Alliance (ICDA) or Open Targets, as well as foundations such as the Lupus Research Alliance, will be important drivers of this vision, as there are many components that are beyond the resources of any given academic or industry lab. I use type 1 interferon signaling as the example, as the pathway is implicated in multiple diseases (see here, here for recent examples in systemic lupus erythematosus [SLE]). Of course, there are many other examples, too (e.g., the IL17A-responsive organoid system described by Nanki et al).
First, catalogue genes in the type 1 interferon pathway (upstream of interferon alpha / beta activation and downstream of interferon signaling). This should be done via annotated gene sets, as well as unbiased approaches such as RNA-sequencing in cells following interferon perturbation. (Note that there are two broad interferon categories, type 1 [interferon alpha and beta], and type 2 [interferon gamma].)
Second, determine which of these genes harbor trait-associated variants related to human disease, including common diseases such as SLE, IBDS or rheumatoid arthritis, and rare immunological diseases such as the interferonopathies and primary immune deficiency. There are many such genes, including those acting upstream of type 1 interferon release (e.g., STING, IFIH1, MYD88, IRAK4, IRF5, TREX1) and downstream of type 1 interferon signaling (e.g., IFNAR2, TYK2, STAT1, STAT2). STAT2 is an interesting example of the allelic series model, as a recent study in Science Immunology described a STAT2 homozygous missense mutation (Arg148Trp), which prevented STAT2-dependent negative regulation of IFNα/β signaling (here). This study builds on other STAT2 variants that are associated with common and rare immunological phenotypes (see here).
Third, determine which of these genes harbor evidence for multiple protein-coding mutations from exome sequencing data of common and rare immunological diseases. This could be done from disease agnostic initiatives such UK Biobank, FinnGen, gnomAD, and East London Genes & Health, or from disease-specific case-control cohorts such as International IBD Genetics Consortium or those previously funded by the Lupus Research Alliance. The most interesting short list of genes would be those for which there are multiple established trait-associated common and/or rare variants and also evidence of association from rare variant burden tests performed with case-control exome sequencing data (even if not yet genome-wide significant). As emphasized above (Section 1A), it is not clear how many trait-associated variants will be discovered from exome sequencing studies.
Fourth, design a high-throughput assay to enable a saturation mutagenesis screen. For type 1 interferon signaling, this could be a system in which the relevant gene has been knocked-out to create a cell line resistant to interferon activation or inteferon signaling. To measure interferon activity (activation or signaling), a reporter construct under control of an interferon-stimulated response element (ISRE) is stably inserted. This ISRE-reporter construct would contain a component to enable high-throughput identification of cells that respond to type 1 interferon signaling (e.g., a marker to enable flow sorting [analogous to CRISPRi-flowFISH] or a toxin that kills cells [analogous to what was done for BRCA1]). Small molecules known to target these gene products could be used to calibrate the assay (see here for TYK2). Individual gene constructs with all possible point mutations could be transfect or engineered into the ISRE-reporter cell line to assay the effect of different point mutations. As emphasized above (Section 2A), the value of a saturation mutagenesis approach is dependent in part upon the number of trait-associated variants expected to be discovered via exome sequencing studies.
Fifth, incorporate functional data into statistical tests of association from exome sequencing data. This step will eliminate those variants that are not functional (and therefore likely not pathogenic), which should in turn improve power to detect true positive associations.
And sixth, test these same variants for pleiotropic effects using PheWAS and related approaches (e.g., TreeWAS) across all available biobanks. For low-frequency and common variants, this will be relatively straightforward; for rare variants, this will be more challenging, as the same rare variants may not be present among individuals not used for the initial discovery; and for ultra-rare variants, as observed in studies of schizophrenia (here), this will not be possible at all.
I want to pose two questions before my conclusion: First, is it better to develop pre-emptively functional assays for all possible protein-coding mutations in a single gene, or restrict functional analysis only to known trait-associated variants from genes within a pathway? The decision is largely dependent on whether you believe there will be a small number (e.g., <10) or a larger number (e.g., >30) of trait-associated alleles per gene. If you believe the former, then one could argue that the most productive effort, with fixed resources, would be to focus on a larger number of genes and small number of observed trait-associated variants within these genes. If you believe the latter, then one could argue that it is more efficient to model all possible mutations in a single gene, as over time many of these will be found to be trait-associated.
Second, what is the ideal assay to assess variant function? While it is possible to use GWAS data to hone in on key biological pathways to design relevant functional assays (here, here), our knowledge of human biology is still in its infancy. Further, a major benefit of unbiased genetic studies is to uncover novel biology, rather than force-fit genetic discoveries into existing biological paradigms. Ideally, functional assays would test every possible allele in every possible cell type under a range of conditions, followed by comprehensive hypothesis-driven functional readouts and hypothesis-free omics profiling. This is important because it is entirely possible that alleles behave differently depending on subtle differences in cellular context. In the absence of such a comprehensive and unbiased approach, it is conceivable that the wrong assays are selected and that function-phenotype curves are generated with the wrong biological information.
In conclusion, I believe that the three assumptions will be proven true in the “clear view” decade to enable the clickable genome. The background and recent published examples provide evidence to support this position. What is less certain is not whether genes will harbor multiple trait-associated alleles but the number of trait-associated alleles per gene. What is also less clear is whether there is sufficient knowledge of human biology to design assays that comprehensively query variant function in a number of different cell types under a range of conditions. Finally, while biobanks will continue to grow, and while new methodologies will be developed to analyze biobank data (e.g., PheCAP, TreeWAS), the rate-limiting steps will be the ability to harmonize and federate data across biobanks. Nonetheless, it seems probable that within the decade these assumptions will be met, which will enable the ability to click on any gene in the genome to generate genetic dose-response curves.