
There were so many good articles and news reports this week on genetics/genomics and drug discovery & development. A few examples include: article in Nature Communications on gene therapy via CRISPR/Cas9 for retinitis pigments (here); a partnership between Editas and Allergan (Matthew Herper story here); Nature Reviews Genetics article by Khera and Kathiresan on genetics of coronary heart disease (here); Genome Magazine article on the importance of pharmacogenetics across ethnic groups to prevent severe adverse events (here); and a victory for pre-prints in challenging the statistical robustness of a publication in Nature Genetics (here).
I decided to focus on a study that provides a mechanistic link between a genetic mutation and a therapeutic hypothesis in Parkinson’s disease. The reason I chose this article is that it highlights the challenges of going from a robust genetic association to a biology hypothesis, and ultimately how to gain confidence in a therapeutic hypothesis with pre-clinical models. As you will see at the end, a clinical trial is now underway to test the therapeutic hypothesis in humans.
The manuscript was published March 7 in PNAS, “Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models” (see here). Scientists at Sanofi/Genzyme performed elegant experiments in two different mouse models to support substrate reduction therapy in the treatment of Parkinson’s disease (PD). Before I summarize the key findings, let me provide some background.
There is a clear link between mutations in glucocerebrosidase (encoded by the GBA gene) and increased risk of PD (here, here). One mutant copy of the GBA gene is required to increase risk of PD five-fold. The same GBA mutations, when inherited in the homozygous state, lead to a lysosomal storage disease, Gaucher’s disease. Consistent with a direct genetic link between PD and Gaucher’s, there is an increased risk for PD among family members related to Gaucher’s patients.
The underlying pathophysiology of Gaucher’s disease is due to accumulation of glucocerebroside in a cells across the body. The disorder is characterized by bruising, fatigue, anemia, low blood platelet count, and enlargement of the liver and spleen – primarily “peripheral” manifestations (i.e., outside of the central nervous system).
There are two effective treatments for Gaucher’s disease: enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). The approved therapies only treat peripheral manifestations of Gaucher’s disease, as neither therapeutic approach effectively penetrates the blood-brain-barrier to get into the central nervous system (CNS). Indeed, Gaucher’s patients treated with either ERT or SRT can go on to develop Parkinson’s disease.
In contrast, the functional link between GBA mutations and risk of PD is less clear (see review here). Two predominant hypotheses by which GBA mutations lead to PD include:
- Substrate hypothesis: loss-of-function (LoF) GBA mutations lead to pathological accumulation of glycosphingolipids [glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph)] in lysosomes, which interfere with normal lysosomal function (e.g., clearance of other proteins such as alpha-synuclein).
- Misfolding hypothesis: Either LoF or gain-of-function (GoF) GBA mutations could lead to misfolding of the GBA protein itself, irrespective of substrate build-up, which could impair lysosomal-mediated alpha-synuclein breakdown and oligomerization. Note that most believe that disease-associated GBA mutations are LoF and not GoF.
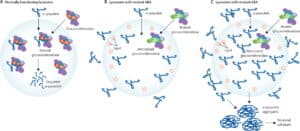
Both hypotheses center on the role of the autophagy-lysosomal pathway, which plays a role in maintaining cellular homeostasis by degrading bulky cytoplasmic material, including damaged organelles and misfolded proteins (see here).
However, the therapeutic implications of the two hypotheses are quite distinct. The Figure below (stolen from here) captures the essential elements of the different approaches in Gaucher’s, and by extension to PD.
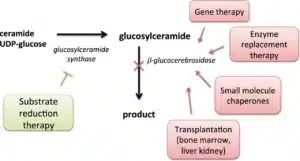
For the “substrate hypothesis”, therapeutic inhibition of the enzyme glucosylceramide synthase (GCS), which catalyzes the synthesis of glucocerebroside, or activation of glucocerebrosidase (GCase) itself, should be effective in treating PD by reducing the accumulation of glycosphingolipids. This is akin to substrate reduction therapy for Gaucher’s disease, as in the Figure above. This hypothesis has been partially tested with miglustat, albeit with mixed results to date (see here). From a genetics perspective, haploinsufficiency of GBA due to complete absence of one of the two copies of GBA supports the substrate hypothesis, as there is no abnormal GBA protein to misfold.
For the “misfolding hypothesis”, it is necessary to clear the mutant GBA protein from the endoplasmic reticulum (ER) and lysosome, thereby preventing dysregulation of these clearance mechanisms. The predominant therapeutic hypothesis is to help GBA traffic through the ER and lysosome, via pharmacological chaperone therapy. I won’t say more here, as the PNAS study focused on substrate reduction therapy via an enzymatic inhibitor of GCS. (For a review of of pharmacological chaperone therapy in PD, see here.)
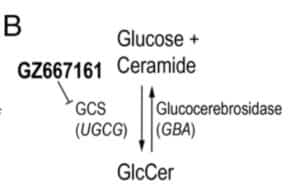
The PNAS study by Sardi et al provides support for the “substrate hypothesis”, as they demonstrate that pharmacological inhibitors of GCS (see Figure 1B, pasted below) have a favorable biochemical and clinical impact in two different mouse models of PD. The study utilized two synucleinopathy murine models: (1) Gba–D409V/D409V, expressing mutant D409V glucocerebrosidase and endogenous alpha-synuclein; and (2) A53T–SNCA model, over-expressing A53T–alpha-synuclein and displaying endogenous wild-type murine glucocerebrosidase.
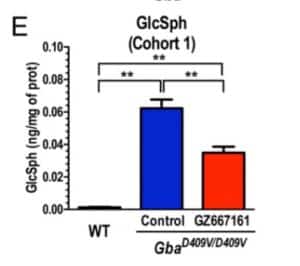
They first showed that a brain penetrant GCS inhibitor, GZ667161, reduces CNS glycosphingolipids relative to untreated mutant mice, although not to the level observed in wild type mice (see Figure 1; also pasted below). The effect was observed in mice treated early (Cohort 1, presymptomatic) and late (Cohort 2, postsymptomatic) in the disease course. These results demonstrate the reduction of glucocerebrosidase substrate glycosphingolipids and confirm CNS target engagement of the GCS inhibitor.
Next, they demonstrated that the GCS inhibitor ameliorates cognitive impairment in the same Gba–D409V/D409V mouse model (see Figure 2). I am not an expert in cognitive mouse models, so it is hard for me to assess the relevance of these findings to humans. However, they did apply two different cognitive assessments, which gives me confidence that the findings are robust.
If the substrate hypothesis is correct, then one would predict that GCS inhibition would lead to clearance of alpha-synuclein aggregates. As shown in Figure 3A (and below), this is exactly what happened: GCS inhibition reduced the accumulation of pathological aggregates of alpha-synuclein.

To extend their findings in a different mouse model with normal GBA activity, they utilized the A53T–SNCA model. As with the Gba–D409V/D409V model, mice with the GCS inhibitor had reduced CNS glycosphingolipid accumulation (Fig. 4A); improved cognitive function (Fig. 4B-C); and reduced membrane-associated and insoluble alpha-synuclein levels (Fig. 4D) and pathological aggregation (Fig. 5). That is, the findings replicate in a separate mouse model of PD.
The study concludes: “In summary, the present findings demonstrate that reducing the levels of CNS glycosphingolipids by antagonizing GCS corrected the hallmark pathological and functional phenotypes in two animal models of synucleinopathy. Glucocerebrosidase deficiency [due to GBA mutations or alpha-synuclein–mediated inhibition can lead to altered glycosphingolipid homeostasis, consequent α-synuclein misprocessing and intracellular deposition, generalized proteinopathy, and the development of behavioral deficits. Reducing the levels of glycosphingolipids through GCS inhibition disrupted the pathogenic cycle of aberrant protein aggregation and functional deficits. Together, the data presented herein provide robust in vivo evidence supporting GCS inhibition as a novel disease-modifying therapeutic approach for GBA-related synucleinopathies, including PD, and support the advancement of GCS antagonists toward clinical testing.”
That is, the data presented by Sardi et al supports the substrate reduction hypothesis: LoF mutations lead to pathological accumulation of glycosphingolipids in lysosomes, which leads to alpha-synuclein aggregation and PD. What is exciting is that this therapeutic hypothesis is now being tested in humans with GZ/SAR402671, a glucosylceramide synthase inhibitor (see clinicaltrials.gov link here).