
Welcome to our first blog of 2015 on genetics/genomics for drug discovery. After a nice vacation in sunny Arizona flying drones (here), I am back soliciting ideas from our Merck Genetic & Pharmacogenomics (GpGx) team. This week’s pick riffs off the events at J.P.Morgan 2015, where there were a number of interesting deals made by pharmaceutical companies and genetic companies (see here, here, here).
With all of this interest in human genetics, it raises the question about how genetics can be used to develop new drugs. The first step is to go from “genes to screens”. That is, the first step is to progress from a human genetic variant associated with a clinical trait of interest to an actual drug screen. This week’s article, published in Nature Chemical Biology, describes one example (see here, here).
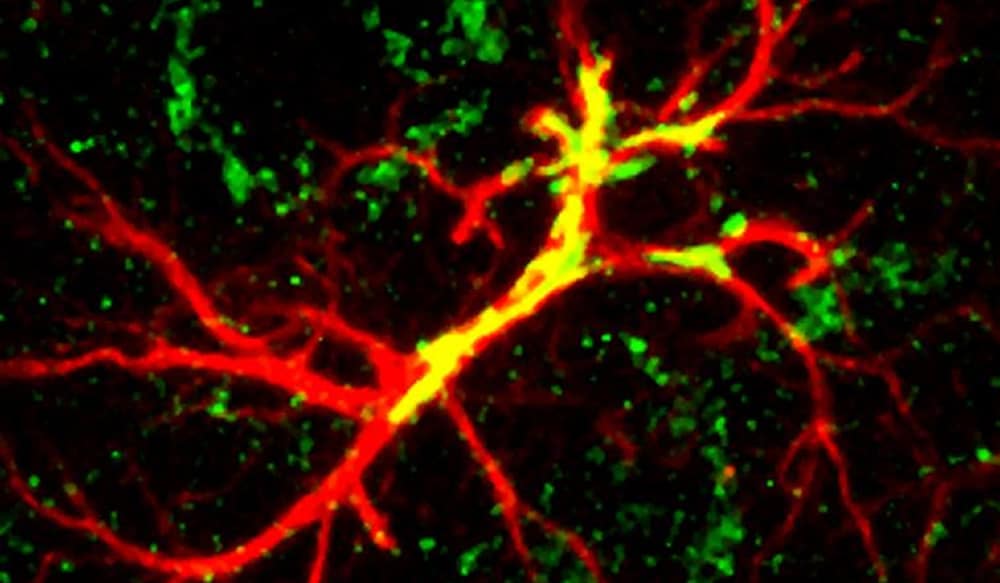
Summary of the manuscript: Deleterious mutations in the ABHD12 gene cause a rare neuroinflammatory-neurodegenerative disorder named polyneuropathy, hearing loss, ataxia, retinitis pigmentosa and cataract (PHARC, see here). A similar phenotype is observed in ABHD12-deficient mice. ABHD12 is an enzyme degrading lysophosphatidylserine (lyso-PS), a signaling lipid known to regulate macrophage activation. The Nature Chemical Biology study by Kamat and colleagues describes the chemical proteomic identification of a related enzyme, ABHD16A, which synthesizes the terminal step leading to lyso-PS generation. A small molecule inhibitor of ABHD16A, which reduces the generation of lyso-PS, was shown to reduce pro-inflammatory cytokine secretion by PHARC-mouse macrophages. Further, the inhibitor normalized the level of lyso-PS in lymphoblast cell lines derived from five PHARC human subjects. Thus, initial human genetics highlighted a neuro-inflammatory pathway and follow-up studies described in this paper might have open the path to a promising target in this pathway for the treatment of PHARC and potentially other neurodegenerative diseases — a category that includes multiple sclerosis, Alzheimer’s, Parkinson’s, ALS and secondary damage after stroke and head injuries.
Why this is interesting: I highlight three interesting aspects of this study. First, this study provides an example where human genetics identifies an important pathway in human disease, endocannabinoid metabolism. The target for small molecule modulation was not the same gene mutated in human disease, but rather a gene (or more precisely, gene protein product) residing in the same pathway. I have commented about the role of pathways in developing therapies anchored in human genetics (see here), and this represents one example.
Second, human genetics raises concerns about other consequences of inhibiting this pathway, which is involved in endocannabinoid metabolism and a wide range of physiological processes (e.g., neurotransmission, mood, appetite, pain appreciation, addiction behavior, and inflammation). Any future drug-mediated interference of this pathway should consider the potential risk of long-term adverse effects.
And third, the study provides an example of how pharmacological tools can be used to understand biological processes. They used a biochemical assay and prior knowledge inhibitors of serine hydrolases to screen a focused small molecule library. They identified a “tool compound” which was used to study the biology of this pathway further.
Thanks to Francois Gervais for his input!
Other articles of interest:
GPR101 mutations responsible for early onset gigantism (NEJM, December 2014). The genetic causes of gigantism in children and acromegaly in adults are poorly understood. In a recent NEJM paper (see here), Dr. Constantine Stratakis and colleagues at the NIH Clinical Research Center reported that a form of early onset childhood gigantism with growth hormone over-production is caused by the duplication of a narrow region on the X chromosome (Xq26.3 microduplication). This locus contains four genes. Through further studies, the authors narrowed the possible causal gene to GPR101 (which encodes a G protein-coupled receptor). Interestingly, the authors then sequenced the implicated gene in samples from 248 adult patients with acromegaly and identified a recurrent GPR101 mutation (E308D) in 11 patients. In vitro studies using a rat pituitary cell line showed that this mutation is an “activating” mutation leading to increased production of growth hormone. The authors proposed that elevation of GPR101 activity, through either gene duplication or gain-of-function point mutation, is responsible for growth hormone overproduction in the majority cases of early onset gigantism in children and a subset of cases of acromegaly in adults. The significance of this study is two folds: (1) it provides clear evidence for a gene-specific genetic screen for a childhood disease and, (2) it sheds light on what controls growth hormone production and may offer insight into additional molecular mechanisms regulating pituitary function and development. Therapeutically, it should be straightforward to develop inhibitors for GPR101 (i.e., GPR101 antagonists) that can be used for treating human patients with gigantism or acromegaly due to overly active GPR101. [Thanks to Richard Chen]
Coronary Heart Disease and Genetic Variants with Low Phospholipase A2 Activity (NEJM, January 2015). Short and sweet! In a brief letter to the Editor (see here), Linda Polfus, Richard Gibbs and Eric Boerwinkle describe a Mendelian randomization experiment for the target of a drug in Phase 3 clinical trials, darapladib (target is lipoprotein-associated phospholipase A2, which is encoded by the PLA2G7 gene). Mendelian randomization is an experimental design in which genetic variation is used to predict the effects of an intervention on risk-factor levels or disease (see previous blog post here). In the current study, they sequenced the exomes of 6,325 participants in the Atherosclerosis Risk in Communities study in search of genetic variants that lower lipoprotein-associated phospholipase A2 activity. Two independent loss-of-function (LOF) variants were associated with lower lipoprotein-associated phospholipase A2 activity, but neither had an effect on cardiovascular-related mortality. The biggest limitation of the study is power, which as the authors state in the Appendix: “A power calculation based on the sample size and genotype frequency in EAs and AAs combined found our study had 74.4% power to detect an effect size 1.73 incident CHD risk.” Nonetheless, the genetic data provides evidence against this target for the reducing the risk of cardiovascular disease. This finding is consistent with the negative clinical trial published last year in NEJM, which was performed on 15,828 patients. [Thanks to Heiko Runz and Dermot Reilly]